

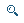
| Аптечное растениеводство | Фармрынки | Фармацевтический маркетинг | Аптекарский огород | Горный чай | Природный лекарь | Травяные сборы |
Телефон: (473) 2605-206
Адрес: г. Воронеж,
ул. Конструкторов, д.31
E-mail: nta@newtechagro.ru
Адрес: г. Воронеж,
ул. Конструкторов, д.31
E-mail: nta@newtechagro.ru
Новости
Статьи
Форум
Полезная информация
Полезные ссылки
Лекарственные растения в народной медицине Диетическое питание Ретро сельский календарь 1980
Статьи
Форум
Полезная информация
Полезные ссылки
Лекарственные растения в народной медицине Диетическое питание Ретро сельский календарь 1980



Доставка сельхозтехники и запасных частей, оросительных систем, насосов во все города России (быстрой почтой и транспортными компаниями), так же через дилерскую сеть: Москва, Владимир, Санкт-Петербург, Саранск, Калуга, Белгород, Брянск, Орел, Курск, Тамбов, Новосибирск, Челябинск, Томск, Омск, Екатеринбург, Ростов-на-Дону, Нижний Новгород, Уфа, Казань, Самара, Пермь, Хабаровск, Волгоград, Иркутск, Красноярск, Новокузнецк, Липецк, Башкирия, Ставрополь, Воронеж, Тюмень, Саратов, Уфа, Татарстан, Оренбург, Краснодар, Кемерово, Тольятти, Рязань, Ижевск, Пенза, Ульяновск, Набережные Челны, Ярославль, Астрахань, Барнаул, Владивосток, Грозный (Чечня), Тула, Крым, Севастополь, Симферополь, в страны СНГ:Киргизия, Казахстан, Узбекистан, Киргизстан, Туркменистан, Ташкент, Азербайджан, Таджикистан.
Результаты научных исследований в области лекарственного растениеводства
Главная > Результаты научных исследований в области лекарственного растениеводства > Химия
Актуальность проблемы и цели реализации программ по лекарственным растениям Анализ динамики заготовок лекарственных трав Действующие ГОСТы на лекарственное сырье История становления фитотерапии. Цели и задачи Организация заготовок и переработка лекарственного сырья на сельских территориях Перспективные технологии выращивания лекарственных растений Рекомендуемая литература Экономика заготовки лекарственного сырья Результаты научных исследований в области лекарственного растениеводства Курортология Целебные свойства пищевых растений Диетическое питание Ретро сельский календарь 1980 Дешевые аналоги лекарств (дженерики) Волшебная палочка здоровья - антисептический биостимулятор Дорогова.
УДК 615.45
В результате хроматографирования суммы лактонов на колонке с «кислой» окисью алюминия при использовании в качестве элементов ряда органических растворителей с возрастающей полярностью были выделены вещества с разными температурами плавления: «A» - l03-105°; «В» - 193-194°; «C» - 82,5-84,5°; «Д» - 350° (с разложением).
В УФ-спектре вещество «A» наблюдается при lmах 230, 248, 258, 332 нм (logε = 4,04; 3,68; 3,59; 4,26). В eго ИК-спектре отмечены следующие полосы поглощения: 1745 см-1 , 1712 см-1(С=0 пиронового кольца и сложно-эфирной группировки), 1630 см-1 ,1585 см-1 (С=0 - ароматической системы). Данные ЯМР-спектра вещества «А» представлены в таблице (CCl4; О-ТМС):

Вышеизложенные данные позволяют приписать веществу «А» структуру рутамарина. Рутамарин из растений отечественной флоры ранее не выделялся, поэтому его идентификация сводилась к установлению структуры.
Вещество «В» по ИК-спектру и пробе смешения идентифицировано с бергаптеном.
Вещества «С» и «Д» по физико-химическим свойствам можно отнести к группе кумаринпроизводных. Химическое изучение их, ввиду малого выхода, нами не проводилось. Кроме того, по данным хроматографии на бумаге «со свидетелем» в корнях Ruta graveolens было отмечено присутствие ксантотоксина.
УДК 07:54
Объектом нашего исследования служили плоды и корни растения, заготовленные в фазу плодоношения. По данным хроматографии на бумаге (бумага Ленинградской фабрики, марки Б, импрегнированная 10% раствором формамида в метаноле; подвижная фаза - н-гексан-бензол-метанол в соотношении 5:4:1; нисходящий метод) в корнях обнаружены кумарины c Rf 0,87 и 0,00, а в плодах - с Rf 0,76 и 0,00.
Лактонную фракцию метанольного экстракта кордей, очищенную от гликозндов, мы хроматографировали на колонке с окисью алюминия. В результате получали хроматографически однородное вещество «А» C19H20P5, стекловидной консистенции, [α]20D -122° (с 0,86; хлорофopм), Rf 0,87, а также вещество «Б» с т.пл. 141 - 143°.
По физико-химическим свойствам вещество «А» явдалось ацилированным кумарином. При его щелочном метанолизе получили оксилактон C14H14О5, т.пл. 178-179°, [α]19D -10,1° (с 0,99; хлороформ), соответствующий по константам 31- окси-31, 41 — дигидроксаетилетину. При анализе ЯМР-спектра вещества «А» было обнаружено два ацильных остатка ангеликовой к сенеционовой кислот; это характеризует выделенное вещество как смесь 31-сенециоилокси- и 31- ангелоилокси - производных 31, 41-дигидросантилетина.
Вещество «Б» по физико-химическим свойствам является стерином.
Из метанольного экстракта плодов растения на колонке с силикагелем был выделен ацилкумарин С19Н20О5, т.пл. 138р-139°, [α]19D 42,5° (с 1,; хлороформ), выход 0,15%, по ИК-спектру и пробе смешения идентифицированный с дигидрофурокумарином - пранчимгином.
Кроме того, выделен стерин с т.пл. 145-147°. По данным хроматографии на бумаге (подвижная фаза - вода-уксусная кислота в соотношении 85:15) в плодах обнаружено наличие трех веществ флавоноидной природы.
УДК 577.1
Селекционные работы, связанные с выведением штаммов спорыньи, продуцирующих алкалоиды определенного состава, изучение вопросов биогенеза и микробиологического синтеза алкалоидов спорыньи требуют точных и быстрых методов количественного и раз¬дельного определения алкалоидов спорыньи.
В настоящем сообщении мы приводим результаты исследований по разработке метода количественного определения эргометрина в рожках спорыньи.
В процессе исследования изучались вопросы экстракции суммы алкалоидов из сырья (растворитель, соотношение растворителя и сырья, время акстракцри), условия разделения суммы алкалоидов методом хроматографии на бумаге и тонком слое сорбента, вопросы десорбции алкалоидов с сорбента (растворитель, время десорбции). Была подобрана система, позволяющая достаточно хорошо разделять алкалоиды.
На основании проведенных исследований разработан метод количественного определения эргометрина. Сущность метода сводится к следующему. Из измельченных рожков спорыньи алкалоиды экстрагируют органическим растворителем в щелочной среде, извлечение сушат сульфатом натрия, определенную часть растворителя отгоняют досуха, остаток растворяют в небольшом объеме спирта. Точный объем этого раствора хроматографируют на бумаге или тонком слое сорбента; светящуюся в УФ-свете зону сорбента, соответствующую эргометрину, снимают и последний десорбируют слабым раствором кислоты, содержание эргометрина в котором определяются фотоколориметрически с реактивом ван-Урка.
Относительная ошибка метода не превышает +5%.
УДК 615.45
По данным ряда исследователей, растения рода Lespedeza обладают диуретическим и гипоазотемическим действием (М.А, Ангарская, В.И. Гудаев, 1971). Во Франции выпускается препарат «Леспенефрил» из леспедеды головчатой ( Lespedeza capitata), обладающий диуретическим и гипоaзотемическим действием. Отечественными и зарубежнымоа исследователями доказано, что диуретический и гипоазотемический эффект леспедепы объясняется наличием флавоноидов. Некоторые виды рассматриваемого рода, произрастающие в Советском Союзе могут, вероятно, представлять интерес для получения препарата с указанным действием.
Нами исследованы флавоноиды четырех видов рода леспедеда. Из исследованных растений выделены и идентифицированы кверцетин (C15H10O7, т.пл. 307 - 312°); кемпферол (C15H10O5, т.пл. 278-281°), апигенин (С15Н20О5, Т.пл. 300°), лютеолин (C15H10O6, Т.ПЛ. 300°), трифолин (C21H20O11•2Н2О, т.пл. 229 - 231°), изокверцитрин (C15H10O6, т.пл. 220 - 222°), гомоориентин (C21H20O11•2Н2О, т.пл. 229 - 231°), ориентин (C21H20O11, •2Н2О, т.пл. 257 - 260°), сапанаретин (C21H20O10, т.пл. 240 - 242°), витексин (C21H20O10, •Н2О т.пл. 250 - 255°), лютеолин-7-глюкозид (C21H20O11, т.пл. 248-250°), леспедин (C27H30O14, т.пл. 189-191°).
Наличие флавоноидов в исследованных водах рода Lespedeza представлено в таблице:
* Сырье для исследования собрал О. В. Журба.
х Флавонолы и их гликозиды ранее были исследованы Г. Г. Задесочной
Первые четыре вида леспедецы содержат вещества флавоноловой я флавоновой природы. Общими для всех видов являются С-гликозиды апигенина и лютеолина (сапонаретин, витексин, гомоориентин, ориентин). Состав же флавоноидов у них варьирует. Так, леспедеца двуцветная содержит в основном вещества флавоноловой природы и незначительное количество С-гликозидов. Это относится и к леспедеце мохнатой, но в отличие от леспедецы двуцветной она содержит значительное количество лютеолин-Т-гликозида и не содержит леспедина. Состав флавоноидов леспедецы копеечниковой отличается от леспедецы даурской лишь отсутствием леспедина.
Леспедеца полосатая существенно отличается от других видов: не имеет веществ флавоноловой природы, а содержит лишь О- и С-гликозиды флавонов.
По составу флавоноидов наиболее близка к леспедеце головчатой (L.capitata ) леспедеца копеечниковая (L. hedysaroides ) и по¬этому ее флавоноиды могут быть рекомендованы для изучения диуретического и гипоазотемического действия.
Леспедеца копеечниковая распространена в Восточной Сибири, Приморском крае и на юге Хабаровского края. Она образует заросли на сухих остепненных склонах и на галечниковых отмелях рек в степных районах. Сырьевая база этого вида достаточно обширна.
УДК 615.45
Репин — бесцветное кристаллическое вещество с т.пл. 154-156°, [α]D20+101°(с 2,0; спирт), состава C19H22O7. Для него предлагаем структуру ацилированного гваянолида (см. рис.).
Углеродный скелет репина был установлен методом дегидрирования как самого репина, так и продукта его гидрирования. При этом был получен углеводород хамазулен. Следовательно, 15 углеродных атомов в репине расположены аналогично хамазулену.
Репин содержит две двойные связи и одну гидроксильную группу, что доказано гидрированием и ацетилированием.
При щелочном гидролизе репина получено два вещества: диоксилактон C15H18О5 и кислота состава С4Н5О3, т.е. репин является сложным эфиром. В ЯМР-спектре кислоты в сильном поле имеется четкий синглет - сигнал метильной группы при углероде, не имеющем хфотонов и связанном с кислородом, а также два дублета по одной протонной единице, относящиеся к сигналам эпоксидных протонов.
При обработке кислоты диазометаном в метаноле получен метиловый эфир. При этом наряду с метилированием произошло раскрытие эпоксида с образованием гликоля. Кислота такого строения не описана в литературе. Мы назвали ее репиновой.
Таким образом, в ЯМР-спектре репина синглет в области 1,5 м.д. и два дублета в области 2,9 и 3,25 м.д. относятся к сигналам ацильного остатка. Квартет в области 4 м.д. является сигналом гемигидроксильного протона, который при ацетилировании, как и следовало ожидать, смещается в область слабых полей. В самом слабом поле находятся сигналы, характерные для протонов экзоциклического метилена, сопряженного с карбонилом γ -лактонного цикла. Уширенные синглеты Б области 5 и 5,25 м.д. относятся к сигналам второго экзоциклического метилена. Гемиацильный протон дает сигнал в области 5,1 м.д.
Большое расстояние между сигналами протонов экзоциклического метилена γ-лактона и структура сигнала H6 указывает на β-положение ацильной группы к метилену γ-лактона. Квартет при 4,7 м.д. - сигнал лактонного протона, который взаимодействует только с двумя протонами; следовательно, лактонный цикл находится в положении С4-С5.
Так как в репине только одна гидроксильная группа и в ИК-спектре отсутствуют полосы поглощения карбонила, кроме γ-лактонного, то мы предположили, что седьмой кислородный атом является эпоксидным. Вторую пару дублетов в области 3,2 и 3,5 м.д. в ЯМР-спектре репина мы отнесли к сигналам протонов эпоксидной группы. Сигналы протонов Н9 и H10 были отнесены с помощью метода ИНДОР. В ЯМР-спектре репина нет сигнала скелетного метила и мы сделали вывод, что пятнадцатый углеродный атом связан с кислородом. То, что эпоксидный мостик связывает первичный и третичный углеродные атомы, доказано следующим образом: при гидролизе репина происходит переориентация эпоксида и освобождение СН2ОН группы, характерные сигналы которой имеются в ЯМР-спектре полученного диоксилактона и его ацетата. Пара дублетов при 3,1 и 3,5 м.д. исчезает и появляется новый сигнал одного эпоксидного протона. При гидрировании диоксилактона происходит гидрогенолиз эпоксида и образуется третичный гидроксид, который не ацетилируется. Третичный гидроксил мог образоваться только в случае, если эпоксидный мостик связан с третичным углеродным атомом.
Неизменность структуры сигнала H10 в ЯМР-спектрах репина и продуктов гидрирования говорит о положении эпоксидного мостика при С3-С5. Константы взаимодействия протонов Н4 и Н5, Н5 и H6, H6 и H10 близки к 10 ГЦ, что указывает на трансположение этих протонов. Для выяснения стереохимии были изучены ЯМР-спектры репина с добавками соли европия, образующей комплексное соединение с гидроксилом. При этом наибольшее смещение наблюдалось для протонов Н9,- Н4, H6. Это говорит о их пространственной близости с гидроксилом, т.е. они расположены по одну сторону углеродного скелета. Относительно меньший сдвиг наблюдался для протонов Н10Н5 и эпоксидных протонов.
УДК 66.095.11
Определение ацетильной группы, как правило, проводят после ее отщепления с помощью соответствующих химических реакций, в результате чего образуется уксусная кислота или ее производные, которые затем определяют различными методами непосредственно в смеси или чаще всего в дистилляте. Однако, если молекула сложная и полифункциональная, в особенности, если в ней присутствуют другие гомологические ацильные группы, то определение ацетильной группы затрудняется или в некоторых случаях требует специфического способа. Кислотный гидролиз проводят в присутствии п-толуол-сульфокислоты, образовывающуюся уксусную кислоту титруют в дистилляте алкалиметрически или иодометрически. Ввиду того, что при этом могут образовываться побочные кислые продукты гидролиза, мешающие определению, принимаются меры по их предупреждению и применяются различные способы для установления поправки.
Щелочной гидролиз отличается от кислотного, как правило, только в первой стадии (гидролиз ОН-). В дальнейшем реакционная смесь подкисляется и определение проводится аналогично.
Кислотный и щелочной алкоголизы ускоряют проведение анализа, однако они также не устраняют помехи, свойственные гидролизу.
Окислительный метод (окисление бихроматом калия по Куну — Роту) ограничен и не может быть использован, если в молекуле имеются С-метильные группы и группы, дающие при окислении уксусную кислоту.
Более совершенными следует считать способы определения ацетильных групп с помощью физико-химических методов.
Метод ядерпо-магнитного резонанса требует навесок вещества до 50 мг (полумикрометод) и, кроме того, некоторые метильные группы имеют близкие химические сдвиги, что существенно затрудняет интерпретацию ЯМР-спектра.
Наиболее совершенными следует считать методы, использующие газовую хроматографию в сочетании с обычными химическими методами отщепления ацетильной группы. При этом образуются метилацетат или свободная уксусная кислота, количество которых расчитывается по площади пика на хроматограмме или автоматически с помощью интегратора.
УДК 615.45
Поскольку все отличие между дигидросамидином и виснадином заключается тодько в адильном радикале, то было бы целесообразно использовать это отличие для разработки метода количественного определения соотношения этих веществ, а суммарное количество делить с помощью УФ-метода. Навеску суммы веществ (20-60 мг ) подвергают щелочному гидролизу 10% NaOH в метаноле в течение 24 часов, затем реакционную смесь подкисляют, а образовавшиеся кислоты экстрагируют эфиром и помещают в хроматограф Xpoм-2 (колонка 3 м х 4 мм, заполнена 20% бегеновой и 0,04% ортофосфорной кислотами на хроматоне NAW HMDS 0,2 - 0,25 мм; ток газа азота во мл/мин.,водорода 60 мл/мин., воздуха 16 мл/мин.; температура колонки 102°). При температуре 102° коэффициент разделения кислот (изовалериановой - дигидросамидин и α-метилмасляной виснадин) равен 1,27, что практически достаточно для кодичественного расчета компонентов до площадям или высотам пиков с относительной точностью ±3%.
УДК 633.88
В результате химической таксации были выявлены перспективные для эксплуатации заросли в субальпийской зоне Месхетского и Триалетского хребтов. В отдельных местообитаниях сырье имело очень высокое содержание платифиллина – от 1 до 3,5 % (при браковочном пределе 0,4 5 на абсолютно сухой вес подземных органов).
Установлены перспективные для эксплуатации местообитаия крестовника в районе Северного Кавказа (Карачаевская и Адыгейская автономные области), в Абхазии по Бзыбскому и Кодоррксому хребтам, в Аджарии близ Годердэского перевала и по Шавштскому хребту. В сырье, собранные в Верхней Сванетии, почти не обнаружено платифиллина. Неустойчивые и колеблющиеся показатели зотифиллина отмечались в крестовнике, выросшем в субальпийской зоне по Рачинркому хребту и в Юго-Осетинской автономной области.
В последние годы заготавливается трава крестовника плосколистного, что позволяет более рационально использовать сырьевую базу в природном ареале. При этом создаются условия для сохранения зарослей от уничтожения.
УДК 615.48
В последнее время внимание наших ученых привлекли три вида кендыря: кендырь коноплевый, к. андрозолистый и к. армянский. Изечением кендыря андрозолистого занимались Ходжаев К.Х., Яматова Р.Ш., Генкина Г.Л., Абубакиров Н.К. (1968 г.). по данным этих авторов, кендырь андрозолистый содержит до 0,74 % суммы сердечных гликозоидов, втом числе более 0,1 % цимарина и 0,3 % К-стpoфaнтина-β.
Во Всесоюзном НИИ лекарственных растений в период с 1967 по 1970 г. проводилось сравнительное сравнительное количественное этих видов кендыря с целью выявления выявления наиболее перспективного источника для получения К-стpoфaнтина-β. Определение содержания суммы сердечных гликозидов, К-стpoфaнтина-β и цимарина в осследуемом сырье (корневище с корнями) проводилось по методике, описанной в литературе (Генкина Г.Л., Шарипов А.Х. и Абубакиров Н.К., 1964 г.).
Полученные результаты показали, что наибольшее количество К-строфантина и цимарина содержится в корнях кендыря конопевого: до 0,44% К-стpoфaнтина-β и 0,32% цимарина (в пересчете на воздушно-сухое сырье). Биологическая активность корней кендыря коноплевого изменяется в пдределах от 120 до 500 ЛЕД на 1 г сырья.
Корни кендыря андрозолистнсго и к.армянского по нашим данным не содержат К-стpoфaнтин-β и цимарин. Биологическая активность коpнeй к.андрозолистного варьирует в пределах от 18 до 66 ЛЕД, кендырь армянский биологически не активен.
Нами была усовершенствована методика количественного определения сердечных гликозидов. Это дало возможность сократить время проведения анализа и использовать на каждое определение значительно меньшую навеску сырья.
УДК 615.45
Нами для анализа смеси папаверина и наркотина предложен метод, основанный на их титровании в неводных средах. Сначала находим суммарное содержание алкалоидов титрованием в ледяной yксусной кислоте. Затем проводим реакцию разрыва лактонного кольца наркотина в среде этанол-бензол-изопропиловый спирт (1:5:4) с помощью спиртового раствора едкого калия и оттитровываем кислотой образовавшиеся продукты реакции. На кривой потенциометрического титрования были получены три скачка: первый соответствовал избытку непрореагировавшей щелочи (по нему рассчитывали содержание наркотина), второй - содержанию карбонатов в щелочи и третий - сумме папаверина и наркотината калия. Параллельно проводам контрольный опыт. Содержание папаверина мы рассчитывали по результатам обоих определений. Титрование проводили на потенциометре ЛП-58 со стандартной парой электродов - стеклянный и каломельный.
КУМАРИНЫ КОРНЕЙ RUTА GRAVEOLENS
В.Б.АНДРИАНОВА
Настойка из корней Ruta graveolens при биологическом испытании показала выраженную антимикробную активность. Для выявления действующих веществ мы исследовали кумарины, содержащиеся в корнях Ruta graveolens, собранных в Ботаническом саду ВИЛР в сентябре 1969 года. Очищенная от фенолов, кислот и других сопутствующих веществ лактонная фракция, по данным хроматографии на бумаге, содержит вещества с Rf 0,00; 0,57; 0,77; 0,89; 0,93 (бумага Ленинградской фабрики марки Б, предварительно импрегнированная раствором 10% формамида в метаноле; подвижная фаза - н-гексан-бензол-метанол в соотношении 5:4:1).В.Б.АНДРИАНОВА
В результате хроматографирования суммы лактонов на колонке с «кислой» окисью алюминия при использовании в качестве элементов ряда органических растворителей с возрастающей полярностью были выделены вещества с разными температурами плавления: «A» - l03-105°; «В» - 193-194°; «C» - 82,5-84,5°; «Д» - 350° (с разложением).
В УФ-спектре вещество «A» наблюдается при lmах 230, 248, 258, 332 нм (logε = 4,04; 3,68; 3,59; 4,26). В eго ИК-спектре отмечены следующие полосы поглощения: 1745 см-1 , 1712 см-1(С=0 пиронового кольца и сложно-эфирной группировки), 1630 см-1 ,1585 см-1 (С=0 - ароматической системы). Данные ЯМР-спектра вещества «А» представлены в таблице (CCl4; О-ТМС):
Вышеизложенные данные позволяют приписать веществу «А» структуру рутамарина. Рутамарин из растений отечественной флоры ранее не выделялся, поэтому его идентификация сводилась к установлению структуры.
Вещество «В» по ИК-спектру и пробе смешения идентифицировано с бергаптеном.
Вещества «С» и «Д» по физико-химическим свойствам можно отнести к группе кумаринпроизводных. Химическое изучение их, ввиду малого выхода, нами не проводилось. Кроме того, по данным хроматографии на бумаге «со свидетелем» в корнях Ruta graveolens было отмечено присутствие ксантотоксина.
УДК 07:54
ХИМИЧЕСКОЕ ИЗУЧЕНИЕ FERULAGO TAURICA
В.В.BAНДЫШЕВ, Л.Г.АВРАМЕНКО
При обследовании зонтичных Крыма на наличие кумаринов было установлено, что Ferulago taurica Schischk. (ферульник крымский) содержит значительное количество этих соединений. Химическому изучению это растение ранее не подвергалось.В.В.BAНДЫШЕВ, Л.Г.АВРАМЕНКО
Объектом нашего исследования служили плоды и корни растения, заготовленные в фазу плодоношения. По данным хроматографии на бумаге (бумага Ленинградской фабрики, марки Б, импрегнированная 10% раствором формамида в метаноле; подвижная фаза - н-гексан-бензол-метанол в соотношении 5:4:1; нисходящий метод) в корнях обнаружены кумарины c Rf 0,87 и 0,00, а в плодах - с Rf 0,76 и 0,00.
Лактонную фракцию метанольного экстракта кордей, очищенную от гликозндов, мы хроматографировали на колонке с окисью алюминия. В результате получали хроматографически однородное вещество «А» C19H20P5, стекловидной консистенции, [α]20D -122° (с 0,86; хлорофopм), Rf 0,87, а также вещество «Б» с т.пл. 141 - 143°.
По физико-химическим свойствам вещество «А» явдалось ацилированным кумарином. При его щелочном метанолизе получили оксилактон C14H14О5, т.пл. 178-179°, [α]19D -10,1° (с 0,99; хлороформ), соответствующий по константам 31- окси-31, 41 — дигидроксаетилетину. При анализе ЯМР-спектра вещества «А» было обнаружено два ацильных остатка ангеликовой к сенеционовой кислот; это характеризует выделенное вещество как смесь 31-сенециоилокси- и 31- ангелоилокси - производных 31, 41-дигидросантилетина.
Вещество «Б» по физико-химическим свойствам является стерином.
Из метанольного экстракта плодов растения на колонке с силикагелем был выделен ацилкумарин С19Н20О5, т.пл. 138р-139°, [α]19D 42,5° (с 1,; хлороформ), выход 0,15%, по ИК-спектру и пробе смешения идентифицированный с дигидрофурокумарином - пранчимгином.
Кроме того, выделен стерин с т.пл. 145-147°. По данным хроматографии на бумаге (подвижная фаза - вода-уксусная кислота в соотношении 85:15) в плодах обнаружено наличие трех веществ флавоноидной природы.
УДК 577.1
О МЕТОДЕ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЭРГОМЕТРИНА В РОЖКАХ СПОРЫНЬИ
Л.Д.ВЕЧКАНОВА
Алкалоиды спорыньи - эрготамин, эрготоксин, эргометрин получили всеобщее признание в медицине. Они применяются в акушерско-гинекологической практике, для лечения сердечно-сосудистой недостаточности, целого ряда заболеваний вегетативной и центральной нервных систем, при лечении болезней пищеварительного тракта, а также в хирургии и дерматологии. Дальнейшее изучение клинических и фармакологических свойств алкалоидов и их производных порождает новые и более интересные области их применения.Л.Д.ВЕЧКАНОВА
Селекционные работы, связанные с выведением штаммов спорыньи, продуцирующих алкалоиды определенного состава, изучение вопросов биогенеза и микробиологического синтеза алкалоидов спорыньи требуют точных и быстрых методов количественного и раз¬дельного определения алкалоидов спорыньи.
В настоящем сообщении мы приводим результаты исследований по разработке метода количественного определения эргометрина в рожках спорыньи.
В процессе исследования изучались вопросы экстракции суммы алкалоидов из сырья (растворитель, соотношение растворителя и сырья, время акстракцри), условия разделения суммы алкалоидов методом хроматографии на бумаге и тонком слое сорбента, вопросы десорбции алкалоидов с сорбента (растворитель, время десорбции). Была подобрана система, позволяющая достаточно хорошо разделять алкалоиды.
На основании проведенных исследований разработан метод количественного определения эргометрина. Сущность метода сводится к следующему. Из измельченных рожков спорыньи алкалоиды экстрагируют органическим растворителем в щелочной среде, извлечение сушат сульфатом натрия, определенную часть растворителя отгоняют досуха, остаток растворяют в небольшом объеме спирта. Точный объем этого раствора хроматографируют на бумаге или тонком слое сорбента; светящуюся в УФ-свете зону сорбента, соответствующую эргометрину, снимают и последний десорбируют слабым раствором кислоты, содержание эргометрина в котором определяются фотоколориметрически с реактивом ван-Урка.
Относительная ошибка метода не превышает +5%.
УДК 615.45
ФЛАВОНОИДЫ РАСТЕНИЙ РОДА LESPEDEZA RICH.
В.И.ГЛЫЗИН
Растения рода Lespedeza. (леспедеда) представлены в Советском Союзе семью видами. Все они произрастают в южной часта Востояной Сибири и Дальнего Востока. В составе рода имеются два вида кустарников, три вида травянистых многолетников и два вида однолетних растений.В.И.ГЛЫЗИН
По данным ряда исследователей, растения рода Lespedeza обладают диуретическим и гипоазотемическим действием (М.А, Ангарская, В.И. Гудаев, 1971). Во Франции выпускается препарат «Леспенефрил» из леспедеды головчатой ( Lespedeza capitata), обладающий диуретическим и гипоaзотемическим действием. Отечественными и зарубежнымоа исследователями доказано, что диуретический и гипоазотемический эффект леспедепы объясняется наличием флавоноидов. Некоторые виды рассматриваемого рода, произрастающие в Советском Союзе могут, вероятно, представлять интерес для получения препарата с указанным действием.
Нами исследованы флавоноиды четырех видов рода леспедеда. Из исследованных растений выделены и идентифицированы кверцетин (C15H10O7, т.пл. 307 - 312°); кемпферол (C15H10O5, т.пл. 278-281°), апигенин (С15Н20О5, Т.пл. 300°), лютеолин (C15H10O6, Т.ПЛ. 300°), трифолин (C21H20O11•2Н2О, т.пл. 229 - 231°), изокверцитрин (C15H10O6, т.пл. 220 - 222°), гомоориентин (C21H20O11•2Н2О, т.пл. 229 - 231°), ориентин (C21H20O11, •2Н2О, т.пл. 257 - 260°), сапанаретин (C21H20O10, т.пл. 240 - 242°), витексин (C21H20O10, •Н2О т.пл. 250 - 255°), лютеолин-7-глюкозид (C21H20O11, т.пл. 248-250°), леспедин (C27H30O14, т.пл. 189-191°).
Наличие флавоноидов в исследованных водах рода Lespedeza представлено в таблице:
| Исследованные растения | Идентифицированные вещества |
| L. bicolor* Л.двуцветная (кустарник) |
Кверцетин, кемпферол, трифолин, изокверцитрин, гомофиентин, ориентин, сапонаретин, витексин, леспедин |
| L, hedysaroides Л. копеечниковая (травянистый многолетник) |
Кверцетин, кемпферол, гомоориентин,ориентин, сапонаретин, витексин, леспедин |
| L. dahurica Л. даурская (травянистый многолетник) |
Кверцетин, кемпферол, апигенин, лютеолин, гомоориентин, ориентин, сапонаретин, витексин |
| L. toraentosa Л. мохнатая (травянистый многолетник) |
Апигенин, лютеолин, гомоориентин, ориентин, сапонаретин, витексин, лютеолин-7-глюкозид |
| L. striata Л. полосатая (однолетнее растение) |
Апигенин, лютеолин, гомоориентин, сапонаретин, лютеоднн-7-глюкозид, (витексин и орнентин – следы) |
х Флавонолы и их гликозиды ранее были исследованы Г. Г. Задесочной
Первые четыре вида леспедецы содержат вещества флавоноловой я флавоновой природы. Общими для всех видов являются С-гликозиды апигенина и лютеолина (сапонаретин, витексин, гомоориентин, ориентин). Состав же флавоноидов у них варьирует. Так, леспедеца двуцветная содержит в основном вещества флавоноловой природы и незначительное количество С-гликозидов. Это относится и к леспедеце мохнатой, но в отличие от леспедецы двуцветной она содержит значительное количество лютеолин-Т-гликозида и не содержит леспедина. Состав флавоноидов леспедецы копеечниковой отличается от леспедецы даурской лишь отсутствием леспедина.
Леспедеца полосатая существенно отличается от других видов: не имеет веществ флавоноловой природы, а содержит лишь О- и С-гликозиды флавонов.
По составу флавоноидов наиболее близка к леспедеце головчатой (L.capitata ) леспедеца копеечниковая (L. hedysaroides ) и по¬этому ее флавоноиды могут быть рекомендованы для изучения диуретического и гипоазотемического действия.
Леспедеца копеечниковая распространена в Восточной Сибири, Приморском крае и на юге Хабаровского края. Она образует заросли на сухих остепненных склонах и на галечниковых отмелях рек в степных районах. Сырьевая база этого вида достаточно обширна.
УДК 615.45
УСТАНОВЛЕНИЕ СТРОЕНИЯ СЕСКВИТЕРПЕНОВОГО ЛАКТОНА РЕПИНА
Р.И. ЕВСТРАТОВА, В.И. ШЕЙЧЕНКО
Из злостного карантинного сорняка горчака ползучего Acroptilon repens L. (DC,), произрастающего в южных районах нашей страны, мы выделили три новых сесквитерпеновых лактона: репин, акроптилин, гирканин.Р.И. ЕВСТРАТОВА, В.И. ШЕЙЧЕНКО
Репин — бесцветное кристаллическое вещество с т.пл. 154-156°, [α]D20+101°(с 2,0; спирт), состава C19H22O7. Для него предлагаем структуру ацилированного гваянолида (см. рис.).
Углеродный скелет репина был установлен методом дегидрирования как самого репина, так и продукта его гидрирования. При этом был получен углеводород хамазулен. Следовательно, 15 углеродных атомов в репине расположены аналогично хамазулену.
Репин содержит две двойные связи и одну гидроксильную группу, что доказано гидрированием и ацетилированием.
При щелочном гидролизе репина получено два вещества: диоксилактон C15H18О5 и кислота состава С4Н5О3, т.е. репин является сложным эфиром. В ЯМР-спектре кислоты в сильном поле имеется четкий синглет - сигнал метильной группы при углероде, не имеющем хфотонов и связанном с кислородом, а также два дублета по одной протонной единице, относящиеся к сигналам эпоксидных протонов.
При обработке кислоты диазометаном в метаноле получен метиловый эфир. При этом наряду с метилированием произошло раскрытие эпоксида с образованием гликоля. Кислота такого строения не описана в литературе. Мы назвали ее репиновой.
Таким образом, в ЯМР-спектре репина синглет в области 1,5 м.д. и два дублета в области 2,9 и 3,25 м.д. относятся к сигналам ацильного остатка. Квартет в области 4 м.д. является сигналом гемигидроксильного протона, который при ацетилировании, как и следовало ожидать, смещается в область слабых полей. В самом слабом поле находятся сигналы, характерные для протонов экзоциклического метилена, сопряженного с карбонилом γ -лактонного цикла. Уширенные синглеты Б области 5 и 5,25 м.д. относятся к сигналам второго экзоциклического метилена. Гемиацильный протон дает сигнал в области 5,1 м.д.
Большое расстояние между сигналами протонов экзоциклического метилена γ-лактона и структура сигнала H6 указывает на β-положение ацильной группы к метилену γ-лактона. Квартет при 4,7 м.д. - сигнал лактонного протона, который взаимодействует только с двумя протонами; следовательно, лактонный цикл находится в положении С4-С5.
Так как в репине только одна гидроксильная группа и в ИК-спектре отсутствуют полосы поглощения карбонила, кроме γ-лактонного, то мы предположили, что седьмой кислородный атом является эпоксидным. Вторую пару дублетов в области 3,2 и 3,5 м.д. в ЯМР-спектре репина мы отнесли к сигналам протонов эпоксидной группы. Сигналы протонов Н9 и H10 были отнесены с помощью метода ИНДОР. В ЯМР-спектре репина нет сигнала скелетного метила и мы сделали вывод, что пятнадцатый углеродный атом связан с кислородом. То, что эпоксидный мостик связывает первичный и третичный углеродные атомы, доказано следующим образом: при гидролизе репина происходит переориентация эпоксида и освобождение СН2ОН группы, характерные сигналы которой имеются в ЯМР-спектре полученного диоксилактона и его ацетата. Пара дублетов при 3,1 и 3,5 м.д. исчезает и появляется новый сигнал одного эпоксидного протона. При гидрировании диоксилактона происходит гидрогенолиз эпоксида и образуется третичный гидроксид, который не ацетилируется. Третичный гидроксил мог образоваться только в случае, если эпоксидный мостик связан с третичным углеродным атомом.
Неизменность структуры сигнала H10 в ЯМР-спектрах репина и продуктов гидрирования говорит о положении эпоксидного мостика при С3-С5. Константы взаимодействия протонов Н4 и Н5, Н5 и H6, H6 и H10 близки к 10 ГЦ, что указывает на трансположение этих протонов. Для выяснения стереохимии были изучены ЯМР-спектры репина с добавками соли европия, образующей комплексное соединение с гидроксилом. При этом наибольшее смещение наблюдалось для протонов Н9,- Н4, H6. Это говорит о их пространственной близости с гидроксилом, т.е. они расположены по одну сторону углеродного скелета. Относительно меньший сдвиг наблюдался для протонов Н10Н5 и эпоксидных протонов.
УДК 66.095.11
МИКРООПРЕДЕЛЕНИЕ АЦЕТИЛЬНЫХ ГРУПП
В.С. КАБАНОВ
В литературе описаны все химические и физико-химические микрометоды определения ацетильных групп с 1929 по 1970 год включительно.В.С. КАБАНОВ
Определение ацетильной группы, как правило, проводят после ее отщепления с помощью соответствующих химических реакций, в результате чего образуется уксусная кислота или ее производные, которые затем определяют различными методами непосредственно в смеси или чаще всего в дистилляте. Однако, если молекула сложная и полифункциональная, в особенности, если в ней присутствуют другие гомологические ацильные группы, то определение ацетильной группы затрудняется или в некоторых случаях требует специфического способа. Кислотный гидролиз проводят в присутствии п-толуол-сульфокислоты, образовывающуюся уксусную кислоту титруют в дистилляте алкалиметрически или иодометрически. Ввиду того, что при этом могут образовываться побочные кислые продукты гидролиза, мешающие определению, принимаются меры по их предупреждению и применяются различные способы для установления поправки.
Щелочной гидролиз отличается от кислотного, как правило, только в первой стадии (гидролиз ОН-). В дальнейшем реакционная смесь подкисляется и определение проводится аналогично.
Кислотный и щелочной алкоголизы ускоряют проведение анализа, однако они также не устраняют помехи, свойственные гидролизу.
Окислительный метод (окисление бихроматом калия по Куну — Роту) ограничен и не может быть использован, если в молекуле имеются С-метильные группы и группы, дающие при окислении уксусную кислоту.
Более совершенными следует считать способы определения ацетильных групп с помощью физико-химических методов.
Метод ядерпо-магнитного резонанса требует навесок вещества до 50 мг (полумикрометод) и, кроме того, некоторые метильные группы имеют близкие химические сдвиги, что существенно затрудняет интерпретацию ЯМР-спектра.
Наиболее совершенными следует считать методы, использующие газовую хроматографию в сочетании с обычными химическими методами отщепления ацетильной группы. При этом образуются метилацетат или свободная уксусная кислота, количество которых расчитывается по площади пика на хроматограмме или автоматически с помощью интегратора.
УДК 615.45
К ВОПРОСУ ГАЗОХРОМАТОГРАФИЧЕСКОГО ОПРЕДЕЛЕНИЯ
СООТНОШЕНИЯ ДИГИДРОСАМИДИНА И ВИСНАДИНА
В ПРЕПАРАТАХ ПО АЦИЛЬНОЙ ГРУППЕ
В.С. КАБАНОВ, В.В.ВАНДЫШЕВ, Б.А.КРИВУТ
Из корней вздутоплодника сибирского подучают препараты димидин и виснадин, действующими веществами которых являются два очень близких по своим химическим и физико-химическим свойствам адидоксикумарина - дигидросамидин и виснадин. На практике чрезвычайно трудно достичь полного разделения этих веществ, и поэтому в препаратах одно из них присутствует в качестве примеси в другом. В настоящее время не существует метода количественного определения отдельно дигидросамидина и виснадина в смеси, так как не было достигнуто полное разделение смеси с помощью колоночной, тонкослойной, бумажной и газо-жидкостной хроматографии. Поэтому УФ-метод дает суммарный результат, а ЯМР-спектр - только грубое соотношение между дигидросамидином и виснадином.СООТНОШЕНИЯ ДИГИДРОСАМИДИНА И ВИСНАДИНА
В ПРЕПАРАТАХ ПО АЦИЛЬНОЙ ГРУППЕ
В.С. КАБАНОВ, В.В.ВАНДЫШЕВ, Б.А.КРИВУТ
Поскольку все отличие между дигидросамидином и виснадином заключается тодько в адильном радикале, то было бы целесообразно использовать это отличие для разработки метода количественного определения соотношения этих веществ, а суммарное количество делить с помощью УФ-метода. Навеску суммы веществ (20-60 мг ) подвергают щелочному гидролизу 10% NaOH в метаноле в течение 24 часов, затем реакционную смесь подкисляют, а образовавшиеся кислоты экстрагируют эфиром и помещают в хроматограф Xpoм-2 (колонка 3 м х 4 мм, заполнена 20% бегеновой и 0,04% ортофосфорной кислотами на хроматоне NAW HMDS 0,2 - 0,25 мм; ток газа азота во мл/мин.,водорода 60 мл/мин., воздуха 16 мл/мин.; температура колонки 102°). При температуре 102° коэффициент разделения кислот (изовалериановой - дигидросамидин и α-метилмасляной виснадин) равен 1,27, что практически достаточно для кодичественного расчета компонентов до площадям или высотам пиков с относительной точностью ±3%.
УДК 633.88
О СОДЕРЖАНИИ ПЛАТИФИЛЛИНА В СЫРЬЕ КРЕСТОВНИКА
ПЛОСКОЛИСТНОГО, СОБРАННОГО В РАЗНЫХ ЗОНАХ АРЕАЛА
Н.М. КАТАМАДЗЕ
Проведенные исследования качества сырья крестовника плосколистного (Senecio platyphуlloides S.et Sev.) из разных зон ареала дозволили установить широкую амплитуду изменчивости содержания платифиллина в нем (от 0 до 3,5%).ПЛОСКОЛИСТНОГО, СОБРАННОГО В РАЗНЫХ ЗОНАХ АРЕАЛА
Н.М. КАТАМАДЗЕ
В результате химической таксации были выявлены перспективные для эксплуатации заросли в субальпийской зоне Месхетского и Триалетского хребтов. В отдельных местообитаниях сырье имело очень высокое содержание платифиллина – от 1 до 3,5 % (при браковочном пределе 0,4 5 на абсолютно сухой вес подземных органов).
Установлены перспективные для эксплуатации местообитаия крестовника в районе Северного Кавказа (Карачаевская и Адыгейская автономные области), в Абхазии по Бзыбскому и Кодоррксому хребтам, в Аджарии близ Годердэского перевала и по Шавштскому хребту. В сырье, собранные в Верхней Сванетии, почти не обнаружено платифиллина. Неустойчивые и колеблющиеся показатели зотифиллина отмечались в крестовнике, выросшем в субальпийской зоне по Рачинркому хребту и в Юго-Осетинской автономной области.
В последние годы заготавливается трава крестовника плосколистного, что позволяет более рационально использовать сырьевую базу в природном ареале. При этом создаются условия для сохранения зарослей от уничтожения.
УДК 615.48
СРАВНИТЕЛЬНАЯ ОЦЕНКА СОДЕРЖАНИЯ К-СTPOФAHTИHA-β
В КЕНДЫРЕ КОНОПЛЕВОМ, АНДРОЗОЛИСТНОМ И АРМЯНСКОМ
В.Н.КУДРЯВЦЕВА
Широко применемый в современной медицинской практике К-стpoфaнтин-β получают в основном из мемян строфанта – растения, семейства кутровых, родиной которых является Восточная Африка. В связи с тем, что строфант представляет собой импортное сырье, возникла потребность в проведении поисков источников К-стpoфaнтина-β во флоре Советского Союза.В КЕНДЫРЕ КОНОПЛЕВОМ, АНДРОЗОЛИСТНОМ И АРМЯНСКОМ
В.Н.КУДРЯВЦЕВА
В последнее время внимание наших ученых привлекли три вида кендыря: кендырь коноплевый, к. андрозолистый и к. армянский. Изечением кендыря андрозолистого занимались Ходжаев К.Х., Яматова Р.Ш., Генкина Г.Л., Абубакиров Н.К. (1968 г.). по данным этих авторов, кендырь андрозолистый содержит до 0,74 % суммы сердечных гликозоидов, втом числе более 0,1 % цимарина и 0,3 % К-стpoфaнтина-β.
Во Всесоюзном НИИ лекарственных растений в период с 1967 по 1970 г. проводилось сравнительное сравнительное количественное этих видов кендыря с целью выявления выявления наиболее перспективного источника для получения К-стpoфaнтина-β. Определение содержания суммы сердечных гликозидов, К-стpoфaнтина-β и цимарина в осследуемом сырье (корневище с корнями) проводилось по методике, описанной в литературе (Генкина Г.Л., Шарипов А.Х. и Абубакиров Н.К., 1964 г.).
Полученные результаты показали, что наибольшее количество К-строфантина и цимарина содержится в корнях кендыря конопевого: до 0,44% К-стpoфaнтина-β и 0,32% цимарина (в пересчете на воздушно-сухое сырье). Биологическая активность корней кендыря коноплевого изменяется в пдределах от 120 до 500 ЛЕД на 1 г сырья.
Корни кендыря андрозолистнсго и к.армянского по нашим данным не содержат К-стpoфaнтин-β и цимарин. Биологическая активность коpнeй к.андрозолистного варьирует в пределах от 18 до 66 ЛЕД, кендырь армянский биологически не активен.
Нами была усовершенствована методика количественного определения сердечных гликозидов. Это дало возможность сократить время проведения анализа и использовать на каждое определение значительно меньшую навеску сырья.
УДК 615.45
ОПРЕДЕЛЕНИЕ ПАПАВЕРИНА И НАРКОТИНА В ИХ СМЕСИ
Т.В.МАКСИМОВА
Наркотин и папаверин, наряду с морфином, кодеином, тебаином и нарцеином, являются главными алкалоидами мака. В процессе разделения алкалоидов мака из опия по методу Грегори папаверин и наркотин переходят в одну фракцию. Описанные в литературе методы количественного определения наркотина и папаверина требуют предварительного разделения двух этих веществ.Т.В.МАКСИМОВА
Нами для анализа смеси папаверина и наркотина предложен метод, основанный на их титровании в неводных средах. Сначала находим суммарное содержание алкалоидов титрованием в ледяной yксусной кислоте. Затем проводим реакцию разрыва лактонного кольца наркотина в среде этанол-бензол-изопропиловый спирт (1:5:4) с помощью спиртового раствора едкого калия и оттитровываем кислотой образовавшиеся продукты реакции. На кривой потенциометрического титрования были получены три скачка: первый соответствовал избытку непрореагировавшей щелочи (по нему рассчитывали содержание наркотина), второй - содержанию карбонатов в щелочи и третий - сумме папаверина и наркотината калия. Параллельно проводам контрольный опыт. Содержание папаверина мы рассчитывали по результатам обоих определений. Титрование проводили на потенциометре ЛП-58 со стандартной парой электродов - стеклянный и каломельный.
Установлено, что завершение реакции нейтрализации в уксусной кислоте можно фиксировать с помощью кислотно-основных индикаторов: генциана фиолетового, бромкрезолового синего, нафтолового красного и бромкрезодового пурпурового. В случае применения обратного титрования вполне пригодными оказались следующие индикаторы: тимоловый синий, параксиленолсульфофталеин, крезоловый красный, ксиленол и галло морская голубая. Первое изменение их окрасок соответствовало оттитровыванию свободной щелочи, второе-сумме папаверина и наркотината калия, что также позволяет вести расчет по этой реакции. Однако в этом случае результаты получаются несколько заниженными в связи с изменяющимся содержанием карбонатов в щелочи. Наряду с этим было найдено, что можно использовать и другие индикaтоpы; фуксин красный, метиленовый голубой, генциан фиолетовый, хинизарин, бромфеноловый синий, бромфеноловый пурпуровый, бриллиантовый желтый и метиленовый красный. Переход окраски при их применении фиксируется только по первому скачку.
Предложенный метод быд опробован на искусственных смесях, соответствующих содержанию алкалоидов в растительных экстрактах. Максимальная относительдая ошибка определения алкалоидов в смеси равна 7%.
УДК 615.45
ТИТРОВАНИЕ НЕКОТОРЫХ ТРИТЕРПЕНОВЫХ И ФЛАВОНОИДНЫХ
ГЛИКОЗИДОВ В НЕВОДНЫХ СРЕДАХ
Т. В. МАКСИМОВА
Как известно, общие химические методы определения тритерпеноыых и флавоноидных гликозидов мало изучены. В связи с этим нами были исследованы условия неводного титрования следующих гликозидов, обладающих биологической активностью: тритерпеноидов – гликозидов олеаноловой кислоты (монозида, биозида и триазида-календулозида В), выделенных из корней календулы, аралозидов А, В и С - из корней аралии маньчжурской, теосапонина - из семян чая, эсдина - из семян конского каштана, а также флавоноида сапонаретина из стручков гледичии.ГЛИКОЗИДОВ В НЕВОДНЫХ СРЕДАХ
Т. В. МАКСИМОВА
В результате этих исследований были разработаны методики количественных опредедений, основанные на кислотно-основном титровании в неводных средах. Учитывая присутствие карбоксильной группы в агликоне (монозид, биозид олеанодовой кислоты) иди в сахарной цепи гликозида (аралозиды, эсцин, теосапонин) и фенольных групп (сапонаретин), мы путем подбора растворителей усиливали кислотные свойства соединений и провшили титрование растворами гидроокиси тетраэтиламмония, едкого кали или натра. Олеаноловую кислоту, ее монозид и биозид титровали в диметилформамиде и метиловом спирте, эсцин и теосапонин - в пиридине. Сапонаретин титровали в среде димeтилфоpмaмидa по двум скачкам, причем в случае титрования спиртовым раствором едкого кали второй скачок значительно возрастает по сравнению с титрованием растворами едкого натра и гидроокиси тетраэтиламмония. Расчет можно проводить по обоим скачкам.
Аралозиды А, В и С мы титровали суммарно в среде метилового или этилового спиртов. Сумму аммонийных солей аралозидов определили обратным титрованием. После добавления метанольного раствора хлористого водорода и последующего титрования pacтвора гидроокиси тетраэтиламмония было три скачка: первый соответствовал оттитровыванию избытка хлористого водорода, второй - освободившейся карбоксильной группе глюкуроновой кислоты и третий - количеству образовавшегося хлорида аммония.
Календулозид В с блокированной карбоксильной группой титровали после предварительного кислотного гидролиза. На кривой потенциометрического титрования было получено два скачка; первый соответствовал избытку метанольного раствора хлористого водорода, второй - освободившейся карбоксильной группе. Титрование проводили при помощи потенциометра ЛП-58 с каломельным электродом сравнения и стеклянным индикaтоpным электродом.
Предлагаемый нами метод быстр и прост в исполнении и дает удовлетворительные результаты. Относительная ошибка определения для всех соединений равна 0,4-2% (среднее из 5-6 определений). С помощью разработанной методики были определены молекулярные веса всех вышеуказанных соединений (максимальная относительная ошибка - ± 5 единиц).
УДК 615.45
КАЧЕСТВЕННЫЙ СОСТАВ ЭКСТРАКТИВНЫХ ВЕЩЕСТВ
3AMAHИХИ ВЫСОКОЙ
О.В.ЖУРБА, И.Н. СОКОЛЬСКИЙ, Н.И. СУПРУНОВ
Для изучения качествевного состава экстрактивных веществ и ях локализадии в отдельных тканях растения нами был проведен хроматографический анализ плодов, листьев, почек, а для корней, стеблей и полегших (укоренившихся стеблей далее именуемых «корневищами») - анализ коры, луба и древесины. Один грамм измельченной ткани экстрагировался 10 мл метанола. В системе петролейный эфирхлороформ (1:l) экстракт разделялся на растворимую липофильную часть и гидрофильную, которая выпадала в осадок. Отфильтрованный осадок был промыт чистой системой растворителей и растворен в метаноле. Смолообразный осадок древесины корней, по-видимому, богат полисахаридами. В экстракте сердпевины стеблей и «корневищ» были обнаружены стерины. Гидрофильные и липофильные вещества подвергались тонкослойной хроматографии в закрепленном слое силикагеля КСК в системах бутанол - этанол —25% аммиак (10:2:5) и хлороформ — метанол (16:1) с последующим проявлением серной кислотой. В качестве свидетеля использовался метанольный раствор биологически активного комплекса заманихи высокой, cocTosnnero из шести веществ тритерпеновой природы, причем три из них менее по- лярны и при проявлении хроматограммы дают фиолетовое окрашива¬ние (Муравьев И.А., Джумаев М.А., 1973).3AMAHИХИ ВЫСОКОЙ
О.В.ЖУРБА, И.Н. СОКОЛЬСКИЙ, Н.И. СУПРУНОВ
Оценка хроматограмм проводилась по цвету пятен и по значению Rf. Гидрофильная часть представлена в основном шестью веществами тритерпеновой природы, которые присутствуют во всех тканях растения, исключая почки и серддевину стеблей и «корневищ», содержащих большей частью липофильные вещества. Серддевина дает на хроматограмме два голубых пятна предположительно стериновой природы. Во всех тканях найдены вещества, относящиеся к фенольным соединениям (одно-два пятна на хроматограмме). Стебли и «корневища» очень близки между собой по составу липофильных веществ, общее число которых достигает десяти. На два вещества меньше имеют корни, листья, серддевина и почки. В плодах обнаружено всего два липофильных вещества.
Более детально биологически активный комплекс стеблей, «корневищ» и корней был изучен при тонкослойной хроматографии (силикагедь Woelm) в системе хлороформ-метанолвода (61:32:7). Проявление пятен проводилось серной кислотой. В качестве свидетелей применялись тритерпеноид лупеол, а также олеоноловая и урсоловая кислоты, не давшие пятен в данной системе. Анализ значения Rf, размера пятен и интенсивнсюти их окраски показал, что качественный и количественный состав комплекса тритерпеноидов в метанольном экстракте стеблей, «корневищ» и корней примерно одинаков. По аначению Rf средний из трех наиболее полярных тритерпеноидав близок к тритерпеноиду лупеолу (см. таблицу).
Значения Rf биологически активного комплекса заманихи высокой
| Объект исследования |
Значения Rf тритерпеноидов | |||||
| 1 |
2 |
3 |
4 |
5 |
6 | |
| Стебли «Корневища» Корни Свидетель-лупеол |
0,427 0,435 0,427 |
0,532 0,548 0,532 |
0,612 0,629 0,612 |
0,798 0,798 0,798 |
0,854 0,862 0,870 0,840 |
0,950 0,950 0,950 |
На основании полученных данных можно констатировать следующее:
- Биологически активный комплекс имеется во всех тканях заманихи высокой (кроме ее сердцевины) и представлен шестью веществами тритерпеновой природы, один из которых близок к тритерденоиду лупеолу.
- Древесина растения богата полисахаридами, которых особенно много в корнях. Сердцевина заманихи содержит стерины. Все растение содержит фенольные соединения. Особенно богата заманиха высокая липофильными веществами, среди которых преобладают жирные и эфирные масла.
- Стебель и «корневище» идентичны по своим качественным показателям. Поэтому стебли заманихи высокой, бесполезно выбрасываемые сейчас при заготовке, целесообразно использовать в качестве сырья, вполне равноценного «корневищам» с корнями.
УДК 615.45
ХИМИЧЕСКОЕ ИЗУЧЕНИЕ КОРНЕЙ И СЕМЯН CORYDALIS ROSEA
Н.Н.МАРГВЕЛАШВИЛИ
Из корней Corydalis rosea beych (сем. Papaveraceae ) дихлорэтановым методом была получена сумма алкалоидов (0,49%), которая затем была разделена на сульфатную фракцию (выход 0,12%) и сумму третичных оснований (выход 0,37%). Из сульфатной фракции выделено оптически неактивное основание состава С20Н13О4 N•Н2О с т.пл. 266-267° (из эфира), не дающее депрессии температуры плавления пробы смешения с сангвинарином.Н.Н.МАРГВЕЛАШВИЛИ
Сумма третичных оснований разделена по основности. Были вы¬делены и идентифицированы пять соединений: протопин С20Н19О5, т. пл. 204-205° (метанол), [α]20DО°; l-аплумидин С20Н17О6 N,т.пл.237° (метанол) [α]20D - 116° (с 1.3 хлф); (d,l-адлумидин С20Н17О6 N, т.пл. 184-186° (метанол), [α]20D О°; l-аплумидин С21Н21О6 N, т.пл. 179-180° (метанол), [α]20D - 42° (с 1.6 хлф); d,l-адлумидин С21Н21О6 N, т.пл. 175° (метанол), [α]20D О°.
После предварительного обезжиривания семян дихлорэтановым методом выделена сумма оснований (1%). В ней отсутствует сангвинарин, остальные же пять присутствуюпшх соединений идентичны алкалоидам, выделенным из корней.
УДК 615.45
МЕТОД КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ГЛАУЦИНА В ТРАВЕ МАЧКА ЖЕЛТОГО
Г. А. MACЛОВА
В связи с внедрением в медицинскую практику гидрохлорида глауцина возникла необходимость в разработке метода количественного определения глауцина в сырье - траве мачка желтого ( Glauci- um flavum С rant z.), который можно было бы использовать для технической документации.Г. А. MACЛОВА
Глауцин является главным алкалоидом надземной части мачка желтого. В литературе есть сведения, что ему сопутствуют более 10 алкалоидов, не считая неидентифицированных оснований, которые усложняют выделение и очистку глауцина.
Нами разработан хроматоколориметрический метод определения глауцина в сырье. Алкалоиды экстрагируют из 5 г сырья органическим растворителем в щелочной среде в соотношении 1:10. 0,1 мл полученного экстракта делят методом ТСХ на отечественном силикагеле КСК (закрепленный слой), используя в качестве подвижной фазы xлоpoфоpм-мeтaнoл-25% аммиак (22,5:2,5:0,1). Пластинку сушат 2 часа на воздухе, проявляют реактивом Драгендорфа, а затем опять сушат 30 минут на воздухе. К снятым с пластинки участкам сорбента с Rf 0,65-0,75, содержащим 100 - 200 мкг глауцина, добавляют 15 мл 1% раствора соляной кислоты и 0,1 мл 1% водного раствора тропеолина 000-2. Тропеолят глауцина экстрагируют хлороформом 2 раза по 15 мл, полученный раствор фильтруют через бумажный фильтр с сернокислым натрием (1 г) в мерную колбу емкостью 50 мл. Через 1 час после добавления тропеотшна измеряют на ФЭК (при 410-420 нм в кювете 1 см) оптическую плотность доведенного до метки xлopoфоpмнoгo раствора, используя в качестве раствора сравнения хлороформ. В области рабочих концентраций поглощение растворов соответствует закону Бера. Относительная ошибка метода не превышает ±5%.
УДК 615.45
ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ ЧЕТВЕРТИЧНЫХ
АЛКАЛОИДОВ САНГВИНАРИНА И ХЕЛЕРИТРИНА
Г.А.МАСЛОВА, О.Е.ЛАССКАЯ
Нами предложен метод неводного потенциометрического титрования четвертичных оснований сангвинарина и хелеритрина, используемых для построеняя калибровочных графиков при раздельном количественном определении сангвинарина и хелеритрина в траве бокконии и в суммарном препарате «сангвиритрин».АЛКАЛОИДОВ САНГВИНАРИНА И ХЕЛЕРИТРИНА
Г.А.МАСЛОВА, О.Е.ЛАССКАЯ
Около 10 г порошка сангвинарина или хелеритрина (точная навеска) растворяют в 20 мл ледяной уксусной кислоты и титруют 0,01 н. раствором хлорной кислоты в уксусной кислоте на потенциометре ЛП-58 со стеклянным и каломельным электродами. Параллельно проводят контрольный отыт. 1 мл 0,01 н. раствора xлоpнoй кислоты соответствует 0,00349 г сангвинарина и 0,00365 г хелеритрина.
Результаты определения приведены в таблице:
| Название | Метрологические данные | ||||
| алкаловда сангвинарин хелеритрнн |
n 5 5 |
Ӿ 91,81 91,76 |
SӾ 0,10 0,21 |
Е95 0,28 0,58 |
Еотн. 0,30 0,63 |
УДК 615.711.6
ИССЛЕДОВАНИЕ АЛКАЛОИДОВ, ВЫДЕЛЕННЫХ ИЗ ТРАВЫ SOPHORA ALOP ECUROIDES
Т.Е.МОНАХОВА
Из травы Sophora alopecuroides (стадия плодоношения) обычным дихлорэтановым методом получена сумма алкалоидов (2,5%). В результате деления по основности и последующего хроматографирования на окиси алюминия IV степени активности были выделены следующие алкалоиды: софоридин (0,37%), C15H24N2O т.пл. 108-110°; баптифолин С15Н20N2O2 (0,0012%), т.пл. 206-208°; цитизин (0,16%), C11H14N2O , т.пл. 151-153°; N - оксиэтилцитизин (0,042%), C13H18N2O2 , т.пл. 63-65°, т.пл. 63 - 65°, [α]D20 - 187°(с 0,725 спирт); основание 4 C15H24N2O2 , Т.ПЛ. 162-164°, [α]D20 - 50,6 (c 0,555 спирт) и основание 6, т.пл. 214-215° - пока не идентифицировано. Цитизин, баптифолин и основание 4 выделены из этого растения впервые. В ИК-спектре основания 4 присутствуют полосы поглоще¬ния: 3620 см-1 (свободный гидроксил), 2800 см-1 ( транс- хиноли- зидин), 1630 см-1 (лактамный карбонил).Т.Е.МОНАХОВА
В масс-спектре имеются пики 264 М+ (78%), 263 (М-1) (100%), 205 (31%), 193 (26%), 166 (37%), М-17 (l0%) и др.
При сравнении масс-спектров основания 4 и матрина можно отметить, что ряд пиков в масс-спектре основания 4 сдвинут на 16 ед. массы, кроме пика 205, который не содержит гидроксила. Следовательно, гидроксил расположен в кольце А.
Данные ЯМР ацетата основания 4 показывают, что рядом с протоном при ацетилированном гидроксиле находятся четыре соседних протона.
На основании вышеизложенного основанию 4 следует приписать структуру З-окси-софоридина:
УДК 615.45
К СТРУКТУРЕ АНДРАХНИНА
Л.П.НАЙДОВИЧ
Андрахнин - алкалоид, выделенный из корней Andrachne rotundifolia. С. А. М. Его элементарный состав С11Н17 NO2 (В.В.Вильямс, Л.П.Найдович, Б.К.Ростодкий, И.А.Губанов, ХПС, №4, 257-260, 1966).Л.П.НАЙДОВИЧ
Учитывая данные элементарного анализа, а также молекулярный вес, равный 356 (определен масс-спектрометрическим методом), для андрахнина была установлена удвоенная формула C21H28N2О3 •Н2О.
Найдено %: С 67, 46; 67, 86; Н 8,73; 8,46; N 7,49; 7,51; ОСН3 17,55; 15,35; Нподв. 0,898. Вычислено %: С 67,38; Н 8,02; N 7,48; (ОСНЗ)216,57; 1,07.
На основании данных ИК-, УФ-, ЯМР-спектроскопии и массспектрометрии для андрахнина можно предположить следующую структурную формулу:
УДК 615.45
ФЛАВОНОВДЫ И ТРИТЕРПЕНОВЫЕ САПОНИНЫ БОБОВ ГЛЕДИЧИИ ЮЖНОЙ
НГО ТХИ БИК ХАЙ, Г.С.ГЛЫЗИНА
Объектом нашего исследования служили бобы гледичии южной, успешно применяемые в народной медицине Вьетнама. В бобах гледичии мы обнаружили значительное количество флавоноидов, тритерпеновых сапонинов и следы алкалоидов.НГО ТХИ БИК ХАЙ, Г.С.ГЛЫЗИНА
Одномерной и двумерной хроматографией на бумаге, качественны¬ми реакциями в исследуемом объекте обнаружено 8 веществ флавоноидной природы.
Хроматографией обезжиренного метанольного экстракта бобов на колонке полиамида с последующей препаративной хроматографией на бумаге нами выделено в индивидуальном состоянии 5 флавонов: лютеолин, C15H10O6, т.пл. 330-332°, λmax C2H5OH 352, 269, 255 нм; ориентин, C21H20O11•H2O, т.пл, 254-257°, [α]D +18° (с 0,46; водный метанол), λmax C2H5OH 351, 268, 256 нм, гомоориентин, C2lH20O11•H2O, т.пл. 228-230°,[α]D +22 (с 0,46; метанол), λmax C2H5OH 352,271,258 нм; сапонаретин, C2lH20O10•H2O, т.пл. 221-223°,[α]D +44 (с 0,7; метанол), λmax C2H5OH 334, 272 нм; витексин, C2lH20O10•H2O, т.пл. 245-247°, [α]D - 13° (с 0,33; метанол), λmax C2H5OH 333, 271 нм. Выделенные флавоны идентифицировали при помощи УФ-, ИК-, ЯМР-спектров.
Хроматографией суммы сапонинов бобов гледичии на колонке с силикагелем выделили новый тритерпеновый гликозид,названный нами аустралозидом.
Аустралозид, С73Н118О39 т.пл. 228 - 231°,[α]D -56° (метанол), м.в. 1618, при кислотном гидролизе дает эхиношистовую кислоту, а в качестве углеводных составляющих - ксилозу, арабинозу, глюкозу и галактозу.
УДК 615.45
ПРЕДВАРИТЕЛЬНАЯ ОЦЕНКА НЕКОТОРЫХ РАСТЕНИЙ
ФЛОРЫ КАВКАЗА НА НАЛИЧИЕ ФЛАВОНОИДНЫХ СОЕДИНЕНИЙ
Д.А.ПАКАЛН, А.М.ЗАХАРОВ, О.И.ЗАХАРОВА, А.Е.ПАКАЛН
В настоящем сообщении представлены данные предварительного качественного химического и хроматографического изучения 317 растительных образцов флоры Кавказа на наличие в них флавоноидных соединений. Обследованные виды относятся к 26 семействам, среди которых наиболее широко представлены растения семейства губоцветных, сложноцветных, бобовых, зверобойных, норичниковых, первоцветных и бурачниковых.ФЛОРЫ КАВКАЗА НА НАЛИЧИЕ ФЛАВОНОИДНЫХ СОЕДИНЕНИЙ
Д.А.ПАКАЛН, А.М.ЗАХАРОВ, О.И.ЗАХАРОВА, А.Е.ПАКАЛН
При качественной оценке использовалась реакция восстановления (Mg + НС1, Zn + HCl ). Хроматографирование проводилось на бумаге марки Ленинградской фабрики №2 им. Володарского в следующих системах: 15% уксусная кислота и бутанол - уксусная кислота - вода (4:1:5). Хроматограммы обрабатывались реактивами, специфичными для флавоноидных соединений.
Полученные данные дают основание рекомендовать ддя углубленного изучения род Primuj.a семейства первоцветных, роды Атеlanchier, Crataegus, Pyrus семейства розоцветных, род Iris семейства касатиковых, род Hypericum семейства зверобойных, роды Ajuga, Calamintha, Dracocephalum, Micromeria, Siderites, Stachys, Teucrium, Ziziphora семейства губоцветных и роды Centaurea, Jnula, Helichrysum, Аnthеmis семейства сложноцветных. Представляют интерес также некоторые роды семейства нсричниковых и бобовых. Мало перспективными для поисков флавоноидных соединений по предварительным данным являются семейства бурачниковых, молочайных и буковых.
УДК 615.45
ХИМИЧЕСКОЕ ИЗУЧЕНИЕ ХИНОНОВ ШАЛФЕЯ ДУБРАВНОГО
Г.Ф.ПРИБЫЛОВА
Шалфей дубравный ( Salvia nemorosa L, ) - полукустарник 30-60 см высотой - произрастает по степям, степным склонам, на суходольных лугах, по опушкам Европейской части СССР.Г.Ф.ПРИБЫЛОВА
Тонкослойной хроматографией на импортном силикагеле марки «G» в системе хлороформ - этилацетат (85:15) было установлено, что в корнях шалфея дубравного содержится не менее шести веществ хиноновой природы, которые были обозначены: №0, №1, №2, N9 3, №4 и №5.
Ранее мы сообщали о выделении и химическом изучении дитерпеноидных хинонов: ацетоксиройлеанона (вещество 1), С22Н30О5, т. пл. 211,5 - 214°, Rf = 0,75,[α]D20 = -12,5° (с = 0,8; CHCl3); оксиройлеанона (вещество 2), С20Н28О4, т.пл. 173-174°, Rf = 0,65, [α]D20 = -130° (с =0,6; CHCI3 ) и неморона (вещество 3), С22Н28О6, т.пл. 192-194° (с разл., из этанола), Rf = 0,50, [α]D20 = +261® (с =0,18; CHCI3).
В значительно меньшем количестве в кjрнях шалфея дубравного находятся вещества №0 и №4, а вещество №5 присутствует только в виде следов.
Вещество №0 состава С20Н28О3, т.пд. 181-183° (блок Кофлера), Rf =0,80, [α]D20 = +134° (с = 0,50; CHCI3) имеет молекулярный вес 316 (масс-спектрометрически). В ИК-спектре вещества №0 (взвесь в вазелиновом масле) имеются полосы поглощения 3340, 1670 (перегиб), 1648, 1628 и 1605 см-1. В хлороформном растворе полосы 1648 и 1628 претерпевают сдвиг и сливаются в одну интенсивную широкую полосу при 1635 см-1, а полоса 3340, относящаяся к оксигруппе, расположенной рядом с карбонилом, смещается до 3390 см-1; полоса 1605 см-1 остается без изменений. Следовательно, в веществе имеются два карбонила, один из которых связан внутримолекулярной водородной связью с соседней оксигруппой. В УФ-спектре вещества №0 имеются λmaxEtOH 204 ( lоg Ɛ 4,29), 270 ( log Ɛ 4,08) и 408 нм (log ε 2,43); λmaxEtOH+NaOH 277 ( log Ɛ 4,37), 270 ( log Ɛ 3,97) и 538 нм (log Ɛ 2,72).
Таким образом, результаты микроанализа на углерод и водород, молекулярный вес, температура плавления, а также данные ИК- и УФ-спектров позволяют идентифицировать вещество №0, С2ОН28О3,с ройлеаноном, ранее выделенным Эдвардсом и сотр. из корней Jnula royleana DC.
Вещество №4, С20Н26О5, т.пл. 195-197° (блок Кофлера), Rf=0,35, = +1360, (с = 0,11; CHCl3) имеет молекулярный вес 346 (масс-спектрометрически). В ИК-спектре вещества №4 (взвесь в вазелиновом масле) имеются полосы поглощения 3550, 3400, 2750, 1714, 1658, 1634, 1618 см-1; в хлороформном растворе отмечены полосы поглощения при 3600, 3420, 2750, 1715, 1655, 1632, 1618 см-1. Полосы 3600 и 3420 см-1 обусловлены оксигруппами, полосы 2750 и 2715 см-1 характерны для колебаний СН и СО связей альдегидной группы; полосы 1655 и 1632 см-1 относятся соответственно первая - к хиноидному карбонилу, вторая - к хиноидному карбонилу, связанному внутримолекулярной водородной связью с соседней оксигруппой; полоса 1618 см-1 обусловлена колебаниями -С = С - связей хинонового кольца. В УФ-спектре вещества №4 имеются λmaxEtOH 205 нм (log Ɛ 4,05), 265 нм (log Ɛ 4,00), 410 нм (log Ɛ 2,85); в щелочной среде наблюдается некоторый сдвиг максимумов поглощения: λmaxEtOH+NaOH 228 нм (log Ɛ 4,20), 265 нм ( log Ɛ 3,80) и 536 нм (log Ɛ 3,10).
При сравнении вещества №4 с продуктом щелочного гидролиза неморона оказалось, что они идентичны, следовательно, вещество № 4 имеет строение 4,4-диметил-7,12-диокси-10-аль-13-изопропил-окта-гидрофенантренхинона-11,14.
УДК 615.45
О СОДЕРЖАНИИ ДУБИЛЬНЫХ ВЕЩЕСТВ В СОПЛОДИЯХ
ТРЕХ ВИДОВ ОЛЬХИ - СЕРОЙ, ЧЕРНОЙ И ОПУШЕННОЙ
З.С.СКРИПНИК
В связи с пересмотром ГОСТа 3851-47 на соплодия ольхи серой (Alnus incana (L) Moench. ) возникла необходимость в изучении содержания дубильных веществ в образцах соплодий, с целью включения этого показателя в проект ГОСТа.
Известно, что вяжущее и кровоостанавливающее свойство соплодий ольхи серой обусловлено значительным содержанием дубильных веществ. Литературные сведения по этому вопросу противоречивы. Одни авторы указывают, что соплодия ольхи содержат значительное количество дубильных веществ, но не приводят их процентное содержание. Другие авторы сообщают, что в соплодиях ольхи содержится незначительное количество дубильных веществ (1,16%), но при этом не указывают вид ольхи.
Нами установлено, что в исследуемых образцах, взятых из товарных партий, содержатся не только соплодия ольхи серой, но и о. клейкой - А. glutinosa ( L.) Gaertn. По литературным сведениям, соплодия ольхи клейкой и о. опушенной - А. hirsuta Turczr используются наравне с о. серой, однако данных о содержании в них дубильных веществ нет. Проведенные нами исследования и были направлены на выяснение и уточнение этих вопросов.
Огфеделение дубильных веществ проведено в 10 средних образцах из товарных партий, полученных от - Украинской и Горьковской товарно-закупочных баз. Дубильные вещества были определены методом, изложенным в Государственной фармакопее СССР X издания, и выражены в процентах в пересчете на абсолютно сухую массу. В этих образцах содержание дубильных веществ колеблется в пределах от 10 до 29% и зависит от места произрастания, времени заготовки и других факторов. Так, образцы соплодий, заготовленные в Закарпатье в 1967 и 1969 гг, имеют 20-29% дубильных веществ, в то время как образцы соплодий, заготовленные в 1967-1969 гг. Горьковской и Украинской базами, содержат их от 10 до 16%.
Кроме того, было определено содержание дубильных веществ в соплодиях ольхи по видам. Образцы соплодий ольхи серой были взяты из товарных партий. Соплодия ольхи клейкой и опушенной были собраны нами осенью 1970 года в ботаническом саду ВИЛР.
В результате полученных аналитических данных выявлено следующее содержание дубильных веществ: в соплодиях ольхи серой от 18 до 34%; в соплодиях о. клейкой от 26 до 30%; в соплодиях о. опушенной от 4 до 6%.
На основании полученных данных нами в действующий ГОСТ, кроме соплодия ольхи серой, рекомендуется ввести дополнительно и ольху клейкую.
Таким образом, нами рекомендованы в действующий ГОСТ 3851-47 следующие дополнения: включен показатель содержания дубильных веществ, что повысит требование к заготовляемому сырью, новый вид ольхи клейкой, что устранит путаницу видов при заготовке сырья и расширит сырьевую базу.
Известно, что вяжущее и кровоостанавливающее свойство соплодий ольхи серой обусловлено значительным содержанием дубильных веществ. Литературные сведения по этому вопросу противоречивы. Одни авторы указывают, что соплодия ольхи содержат значительное количество дубильных веществ, но не приводят их процентное содержание. Другие авторы сообщают, что в соплодиях ольхи содержится незначительное количество дубильных веществ (1,16%), но при этом не указывают вид ольхи.
Нами установлено, что в исследуемых образцах, взятых из товарных партий, содержатся не только соплодия ольхи серой, но и о. клейкой - А. glutinosa ( L.) Gaertn. По литературным сведениям, соплодия ольхи клейкой и о. опушенной - А. hirsuta Turczr используются наравне с о. серой, однако данных о содержании в них дубильных веществ нет. Проведенные нами исследования и были направлены на выяснение и уточнение этих вопросов.
Огфеделение дубильных веществ проведено в 10 средних образцах из товарных партий, полученных от - Украинской и Горьковской товарно-закупочных баз. Дубильные вещества были определены методом, изложенным в Государственной фармакопее СССР X издания, и выражены в процентах в пересчете на абсолютно сухую массу. В этих образцах содержание дубильных веществ колеблется в пределах от 10 до 29% и зависит от места произрастания, времени заготовки и других факторов. Так, образцы соплодий, заготовленные в Закарпатье в 1967 и 1969 гг, имеют 20-29% дубильных веществ, в то время как образцы соплодий, заготовленные в 1967-1969 гг. Горьковской и Украинской базами, содержат их от 10 до 16%.
Кроме того, было определено содержание дубильных веществ в соплодиях ольхи по видам. Образцы соплодий ольхи серой были взяты из товарных партий. Соплодия ольхи клейкой и опушенной были собраны нами осенью 1970 года в ботаническом саду ВИЛР.
В результате полученных аналитических данных выявлено следующее содержание дубильных веществ: в соплодиях ольхи серой от 18 до 34%; в соплодиях о. клейкой от 26 до 30%; в соплодиях о. опушенной от 4 до 6%.
На основании полученных данных нами в действующий ГОСТ, кроме соплодия ольхи серой, рекомендуется ввести дополнительно и ольху клейкую.
Таким образом, нами рекомендованы в действующий ГОСТ 3851-47 следующие дополнения: включен показатель содержания дубильных веществ, что повысит требование к заготовляемому сырью, новый вид ольхи клейкой, что устранит путаницу видов при заготовке сырья и расширит сырьевую базу.
УДК 615.45
ХИМИЧЕСКИЙ СОСТАВ КОРНЕЙ ДУДНИКА ТРОЙЧАТОГО
А.И.СОКОЛОВА, М.Г.ПИМЕНОВ
Для химического изучения дудника тройчатого (Angelica ternata RgL et Schmalh.) были использованы образцы сырья, собранные на озере Искандеркуль в Средней Азии. Из метанольного экстракта корней растения методом хроматографии на силикагеле неполярной фракции выделены три вещества: вещество 1, С26Н52О2, т.пл. 71-73°; вещество 2, С29Н50О, т.пл. 139-140° и вещество 3, С10Н10О4 т.пл. 173-174,5°.А.И.СОКОЛОВА, М.Г.ПИМЕНОВ
Вещество 1 по физико-химическим константам соответствует гексакозановой кислоте.
Вещество 2 на основании ИК—спектра и пробы смешения идентифицировано с β-ситостерином.
Вещество 3 по данным ИК- и ЯМР-спектроскопии является 3-метокси-4-оксикоричной (феруловой) кислотой. Последняя выделена также при хроматографировании на силикагеле продукта, полученного экстракцией водной фазы смесью хлороформ-спирт (1:1).
Из сгущенной водной фракции выделено вещество 4, т.пл. 187-188°, идентифицированное по пробе смешения и значению Rf в тонком слое в присутствии «свидетеля» с сахарозой.
Кумарины в растении не обнаружены.
УДК 615.45
ТРИТЕРПЕНОВЫЕ ГЛИКОЗИДЫ (САПОНИНЫ)
И.Н.СОКОЛЬСКИЙ
Интенсивное исследование природных соединений в последние годы обусловлено, прежде всего, широким применением современных физико-химических методов. При этом большое внимание уделяется выделению и химическому изучению тритерпеновых гликозидов. В настоящем обзоре приводятся данные о выделении, очистке, идентификации и установлении структуры тритерпеновых сапонинов, при этом рассматривается группа гликозидов, агликонами которых являются высокогидроксилированные производные β-амирина, этерифицированные жирными и ароматическими кислотами.И.Н.СОКОЛЬСКИЙ
Полное установление структуры, включающее в себя определение строения агликша, углеводной и кислотной части, места и способа присоединения углеводного и кислотного компонента к агликону, проведено лишь для небольшого числа соединений подобного типа. Наиболее полно, с использованием методов ЯМР-спектроскопии, изучены агликоны. Выяснение структуры гликозида затруднено тем, что три- терпеновые гликозиды в растениях часто представлены смесью, состоящей из большого числа близких по строению веществ, получить которые в индивидуальном виде весьма трудно. Разделение на отдельные компоненты проще проводить у сапонинов с простым агликоном (олеаноловая кислота, хедерагенин и т.д.), чем у сапонинов, в основе котсрых лежит высокогидроксилированный агликон, этерифи- цированный самыми разнообразными кислотами. Достичь разделения последних практически весьма трудно. Так, из семян Tea sinen¬sis L. мы выделили кристаллический гликозид, отвечающий общепринятым критериям индивидуальности, состава С59Н90О27 • Н2О, т. пл. 222-224° [α]D19 +10° (С 2,7; 70% этанол), М. 1285 (эбулиоскопически в этаноле), в углеводную часть которого входят глюкуроновая кислота, галактоза, арабиноза и ксилоза в соотношении 1:1:1:1. Агликоны представлены теасапогенолами А и Е, дигидроприверогенином А, камеллиагенином D и баррингтогенолом С. Кислотная часть состоит из уксусной, ангеликовой и тиглиновой кислот. Очевидно, что в данном случае мы имеем дело с чрезвычайно сложной смесью сапонинов. Для выяснения структуры каждого из них должна быть привлечена статистическая оценка, опирающаяся на наличие соединений с общим принципом строения и позволяющая, на основе фрагментов с выясненной структурой, делать вывод об общей структуре отдельного гликозида.
ИНЖИР КАК ИСТОЧНИК ПРОИЗВОДСТВА НОВОГО ПРЕПАРАТА «ФУРАЛЕН»
Э.А.ЯРОШ
Инжир обыкновенный (Ficus carica L.) семейства тутовых (Moraceae ) занимает видное место среди плодовых культур Закавказья. Кроме культурных сортовых насаждений, в отдельных местах Закавказья имеются природные заросли одичавших деревьев. В инжире обнаружены биологически активные соединения из группы фурокумаринов, поэтому нами были проведены фитохимические исследования инжиров Закавказья с целью обоснования их комплексного использования для нужд медицины.Э.А.ЯРОШ
При исследовании ссртовых и одичавших инжиров Грузии было установлено, что во всех частях растения (в листьях, коре стеблей и корней, в соплодиях) находятся два наиболее активных фурокумарина — псорален и бергаптен. В высушенных листьях, собранных в летне-осенний период, псоралена содержится 0,28-0,49%, бергаптена -0,03-0,1%.
Впервые из листьев инжира, произрастающего в Закавказье, было выделено кристаллическое вещество состава С17Н18О9, которое на осноавании ИК- и ЯМР-спектроскопии элементарного состава, физико-химических свойств и по отсутствию депрессии температуры плавления в пробах смешения идентифицировано как глюкоаид фурокумариновой кислоты. Выход 0,11%.
Определена протеолитическая активность сока из листьев и зеленых стеблей садовых и одичавших деревьев инжира в разные периоды вегетации.
Из семян инжира было выделено, а затем исследовано жирное масло (выход составляет 29-30%).
В итоге был разработан лабораторный регламент получения из листьев препарата «фурален», предназначенного для лечения кожного заболевания витилиг. В фурален входит смесь двух фурокумаринов - псоралена и бергаптена с преобладанием (70-80%) первого. Выход из сырья составляет 0,15-0,20%. Препарат разрешен Фармакологическим комитетом Минздрава СССР для клинических испытаний.
УДК 615.45
О РАЗДЕЛЕНИИ ОПИЙНЫХ АЛКАЛОИДОВ
С ПОМОЩЬЮ ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ
ПРИ КОЛИЧЕСТВЕННОМ АНАЛИЗЕ СЫРЬЯ
А.В.ГАЕВСКИЙ
Тонкослойной хроматографии опийных алкалоидов посвящено большое количество работ, однако известные способы зачастую пригодны лишь для качественной или полуколичественной оценки сырья. Как правило, при ХТС пятна побочных оснований (лауданидина, котарнолина, наркотолина и других) неудовлетворительно отделяются от главных компонентов и мешают элюированию последних. Это особенно характерно для экстрактов из коробочек масличного мака, где уровень ряда сопутствующих соединений может превышать содержание главных алкалоидов (кодеина, тебаина, папаверина, наркотина). Методы хроматографирования на силикагеле КСК (Степаненко, Шемякин, 1970; Чичиро и др., 1971) не позволяют надежно отделять наркотин от папаверина. Быстрые денситометрические иди фотометрические способы оценки хроматограмм (без элюирования компонентов) пока еще малопригодны для количественнсяго анализа сырья, даже при использовании стандартной аппаратуры. Колориметрический метод на основе ХТС с элюированием, разработанный для кодеина и тебаина (Дечеи, Жадон, 1972), громоздок и неудобен при серийных определениях. Практически отсутствуют методы разделения неочищенных экстрактов из коробочек масличного мака при быстром термическом извлечении алкалоидов.С ПОМОЩЬЮ ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ
ПРИ КОЛИЧЕСТВЕННОМ АНАЛИЗЕ СЫРЬЯ
А.В.ГАЕВСКИЙ
В результате испытания различных способов подготовки сорбента и многочисленных подвижных фаз (известных и оригинальных) нами выявлены условия хроматографирования 5 главных алкалоидов в коробочках масличного мака и 4 нефенольных алкадоидов в опии-сырце, обеспечивающие возможность последующего элюирования компонентов к их количественного определення колориметрическим (морфин, тебаин) и УФ-спектрофотометрическим (кодеин, папаверин, наркотин) способом.
При анализе морфина в коробочках мака неочищенные экстракты хроматографируют на нейтральном силикагеле КСК в смеси хлороформа с изопропидовым спиртом и аммиаком (30:10:1); морфин элюируют 0,1 н. раствором соляной кислоты и определяют на колориметре (при 435 нм) или спектрофотометре (при 450 нм) по известной цветной реакции с нитритом натрия и аммиаком.
При определении нефенольных алкалоидов экстракты из опия и коробочек масличного мака очищают от фенольных оснований и балластных веществ и хроматографируют на щелочном силикагеле КСК в смеси хлороформа с ацетоном и метанолом (7:2:1) — для кодеина и тебаина — и в смеси этилацетата с бензолом (3:1) - для папаверина и наркотина.
Кодеин элюируют метанолом и определяют спектрофотометрически при 225 нм; тебаин элюируют 1% раствором соляной кислоты (с одновременным гидролизом на кипящей бане) и определяют в условиях, разработанных для морфина. Папаверин и наркотин элюируют смесью хлороформа со спиртом и концентрированным аммиаком (90:10:1,5) и определяют (после растворения в 0,1 н. соляной кислоте) спектрофотометрически при 225 или 312 нм (наркотин) и при 250 нм (папаверин).
Селективность применяемых подвижных фаз проверена двумерной хроматографией, повторным хроматографированием на силикагелях различных марок и на бумаге, а также (для морфина) сопоставлением результатов анализа сырья разработанным методом и известным специфичным бумажно-хроматографическим способом ХНИХФИ.
Подлинность компонентов подтверждена данными ИК-спектроскопии после выделения с помощью препаративной ХТС в испытуемых условиях.
Относительная погрешность среднего результата (Sx) определения морфина в коробочках мака при n =8 составила +2,3%.
Ошибки среднего результата определения нефенольных алкалоидов в опие: для кодеина ±4-5%, тебаина ±3-4% (при n = 6), папаверина ±2,7%, наркотина ±2,8% (при n=8); в коробочках мака ошибка определения тебаина составила ±4-5%, а для других нефенольных компонентов, при низком их содержании, возросла до 6-8%.
Оказалось возможным проводить анализ всех 4 алкалоидов в коробочках мака после хроматографировании на одной пластинке в условиях, принятых для разделения кодеина и тебаина. В этом случае элюируют суммарное пятно папаверина и наркотина и определяют компоненты (при 225 и 250 нм) известным спектрофотометрическим методом (Суранова, 1972) с относительной ошибкой ±8,4-9,4%.
Несмотря на существенный разброс показаний, метод вполне пригоден для оценки сырья на заключительных стадиях селекционного процесса, поскольку межссртовые различия намного превышают относительную ошибку анализа.
Предложен способ определения хроматографической чистоты главных алкалоидов мака на силикагеле с помощью 3 подвижных фаз различной полярности, например, сочетания двумерной хроматографии в смесях дихлорэтан-метанол-20% раствор аммиака (50:30:10:3) и этилацетат-бензол-ацетонитрил-25% растворе аммиака (50:30:15:2) с обратным (от финиша к старту) хроматографированием в чистом этилацетате. При разделении указанным способом суммы алкалоидов в очищенных экстрактах из коробочек мака в дозах, соответствующих 200-250 мг сырья, обнаружено свыше 30 компонентов, окрашиваемых реактивом Драгендорфа (по-видимому, нативных соединений и артефактов).
Для одномерного разделения 5 главных алкалоидов (полуколичественный анализ) наиболее пригодными оказались некоторые «среднеполярные» аммиачные смеси растворителей, в первую очередь ацетон-толуол-этанол-25% раствор аммиака (50:40:6:2) на щелочном силикагеле. Обнаружено, что добавление аммиака в жидкую фазу неравнозначно подщелачиванию cорбентa 1% раствором карбоната натрия или 0,15-0,20 н. раствором едкого кали, и оптимальный эффект разделения опийных алкадоидов может быть достигнут при сочетании этих приемов.
Проведена хроматографическая проверка объемного метода (Дьердь, 364), применявшегося ранее для определения нефенольных алкалоидов в селекционных образцах мака (Шаркани и др., 1965,1966). Установлено, что объемный метод, как правило, завышает результаты, по сравнению с хроматографией, из-за присутствия побочных соединений во фракциях оснований средней силы (кодеин + тебаин) и папаверина.
УДК 615.45
ГАЗОХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
ФУРОКУМАРИНОВ В ПРЕПАРАТЕ «ФУРАЛЕН»
Б.А.КРИВУТ, В.С.КАБАНОВ, Н.А.ФЕДЮНИНА
В литературе известны спектрофотометрические методы определения фурокумаринов псоралена и бергаптена в препаратах «фурален» и «псоберан». Один из них - с предварительным хроматографическим разделением и другой без разделения суммы фурокумаринов.ФУРОКУМАРИНОВ В ПРЕПАРАТЕ «ФУРАЛЕН»
Б.А.КРИВУТ, В.С.КАБАНОВ, Н.А.ФЕДЮНИНА
Нами разработан метод количественного определения фурокумаринов в препарате «фурален» с применением газо-жидкостной хроматографии.
Разделение псоралена и бергаптена осуществлялось на стеклянной колонке (длина 120 см и вн. диаметр 3 мм) заполненной 0,6% полиэтиленгликольадипинатом на хромосорбе AW 60-80 меш. Хроматограф – «Хром-4», детектор - ПИД. Расход газов: азота -50мл/мин, водорода - 35 мл/мин и воздуха - 500 мл/мин; температура колонки - 180° С, испарителя - 200°. Доза - 2 мл 1% раствора фуралена в этаноле. Время выхода псоралета 2,3 мин., бергаптена - 5,8 мин., время полного анализа ~ 7 минут. (рис. 1).
Так как препарат «фурален» состоит только из двух фурокумаринов, процентное содержание псоралена и боргаптена можно определить по отношению доли одного компонента к сумме компонентов, которая принимается за 100%.

Рис. Хроматограмма препарата «фурален».
Относительное содержание псоралена (или бергаптена) равно высоте соответствующего пика, умноженной на его время удержания и умноженной на поправочный коэффициент.
Содержание фурокумаринов рассчитывают по следующим формулам:
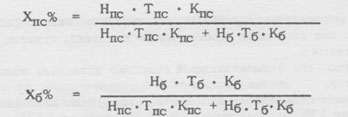
где: Hпс и Hб - соответствующая высота пика псоралена и бергаптена, мм;
Тпс и Tб - соответствующее время удержания псоралена н бергаптена;
Кпс и Kб - соответствующий поправочный коэффициент для псоралена = 1,014, для бергаптена = 1,082.
Поправочные коэффициенты для псоралена и бергаптена определяли на искусственных смесях.
Разработанный метод проверен на 5 сериях препарата «фурален». Результаты анализов совпали с данными, подученными по известному спектрофотометрическому методу.
Сравнительные данные приведены в таблице.
Таблица. Результаты сравнения спектрофотометрического и газометричеекого методов определения псоралена и бергаптена в препарате «фурален».
| № серии |
Соединение | Спектрофотометрнческий метод, % |
Газохроматографический метод, % |
Разница определений, % |
| 1 2 3 4 5 1 2 3 4 5 |
псорален бергаптен |
83,69 82,01 84,75 84,69 84,14 18,21 15,69 14,75 14,81 15,66 |
82,79 82,16 82,56 83,41 82,94 17,60 15,24 14,90 14,73 15,84 |
1,08 0,18 2,65 1,53 1,45 3,47 2,96 1,01 0,54 1,14 |
УДК 615.45
МИКРОБИОЛОГИЧЕСКИЙ ГИДРОЛИЗ ГЛИКОЗИДОВ ALLIUM ANQULOSUM
А.Ф.АЗАРКОВА, В.С.ФОНИН, Л. М. КОГАН
Мы исследовали возможность использования микробиологическсго гидролиза для получения диогенина из соцветий, луковиц и корней лука угловатого.А.Ф.АЗАРКОВА, В.С.ФОНИН, Л. М. КОГАН
Известно, что ферментативный гидролиз позволяет повысить выход генинов.
Проверена способность культур Аspergillus terreus 388 В, Asp.niger 136В, Asp.niger 135В, Aspniger 137В, Asp.oryzae - 283В (культуры грибов из музея ВНИИА любезно предоставлены В.Д. Кузнецовым) гидролизовать гликозиды без их выделения из растения. 5 г измельченных соцветий, луковиц или корней лука угловатого (сырье собрано и предоставлено Т.М.Мельниковой) суспензировали в 100 мл водопроводной воды, содержащей 200 мг NН4NО3 b 1 г СаСО3 и инокулировали культурой гриба, выращенного на питательной среде с кукурузным экстрактом. Ферментацию проводили на качалке при 28°, периодически отбирая пробы. После экстракции и обработки пробы анализировали ТСХ на силикагеле КСК з системе циклогексан-этнлацстет (4:1) для генинов и в системе хлороформ-метанол-вода (65:35:10) для гликозидов. Предварительное обезжиривание лука хлороформом несколько ускоряет гидролиз гликозидов аспергиллами, а экстракт в этом случае содержит меньше балластных веществ.
Все испытуемые штаммы гидролизу ют гликозиды в соцветиях, луковицах и корнях Лука угловатого с образованием диосгенина, причем максимальный выход диосгенина достигается на 3-5 суток ферментации. При ферментации не происходит образование 3,5—спиростадиена. Наиболее высокий выход диосгенина достигнут (по данным ТСХ) при гидролизе культурами Asp. terreus 388В, Asp. niger 136В, Asp.niger 137В. В случае Asp.niger 137В. образуется меньше метаболитов, затрудняющих выделение диосгенина, и этот штамм был отобран для более глубокого изучения.
В процессе ферментации постепенно исчезают исходные гликозиды и появляются менее полярные вторичные гликозиды с предположительно меньшим числом сахаров в углеводной цепи и диосгенин, т.е, идет ступенчатый гидролиз.
Для препаративного получения диосгенина 50 г измельченных обезжиренных соцветий ферментировали 5 суток с культурой Азр. niger 137 В в указанных условиях. Ферментационную массу фильтровали, осадок промывали водой, сушили при 60° и экстрагировали хлороформом в аппарате Сокслета. Экстракт вьшаривали и остаток хроматографировали на колонке с силикагелем, вымывая смесью циклагексан-этилацетат (4:1) диосгенин, т.пл. 202-5° (ацетон), выход 0,21% (считая на абсолютно сухое сырье), идентичный заведомому образцу.
УДК 615.45
ИССЛЕДОВАНИЕ СТОЙКОСТИ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ
В ПРОИЗВОДСТВЕННЫХ УСЛОВИЯХ
Проф., д-р Е.ЛЮТОМСКИЙ
Институт растительно-лекарственной промышленности, г. Познань.ПРЕПАРАТОВ РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ
В ПРОИЗВОДСТВЕННЫХ УСЛОВИЯХ
Проф., д-р Е.ЛЮТОМСКИЙ
В отличие от теоретических поисков, исследования стойкости (устойчивости) лекарственных форм препаратов в фазе технологической разработки для ну лсд производства должны быть до некоторой степени упрощены. Из ряда проблем следует выдвинуть прежде всего только наиболее важные, имеющие практическое значение в области использования препаратов. При исследовании стойкости лекарственных средств как продукта промышленного производства следует учитывать требования технологического характера. При разработке технологических требований следует иметь в виду получение высококачественного препарата, обладающего высокой биологической активностью. Для этого необходимо подобрать оптимальное рН раствора и наиболее эффективное консервирующее вещество.
Долгое время господствовало мнение, что правильно разработанный метод анализа - это основное, что обеспечивает доброкачественность лекарственного средства. Самым важным считался качественный состав препарата. Многочисленные критические замечания при обсуждении этого вопроса, указывающие на недостаточность качественных норм, заставили ведомство фармацевтического надзора заинтересоваться самим процессом производства лекарственного препарата, так как отмечались случаи, когда лекарственное средство не обладало должным качеством вследствие неправильного изготовления или вследствие использования недоброкачественного сырья.
Обмен взглядами, проведенный в конце 1968 года в Хельсинках на конференции Международной Организации по охране здоровья, показал достаточно ясно, что проблема контроля процесса производства лечебных препаратов рассматривается всюду как важный и актуальный вопрос.
Определение содержания биологически активных веществ представляет собой решающий критерий для оценки стойкости (устойчивости) препаратов. Однако применяемый аналитический метод должен отвечать следующим требованиям:
- он должен быть сравнительно простым и пригоден для серийных определений;
- он должен быть настолько чувствителен, чтобы с его помощью можно было определять незначительные количества вещества, например, в сильно разбавленных растворах;
- точность метода должна быть такова, чтобы это обеспечивало возможность точного определения стойкости препарата;
- наиболее удобным является метод, позволяющий определить биологически активное вещество и продукты его разложения. Наиболее пригодными для этой дели является спектрографические методы в УФ области с предварительным хроматографическим разделением.
Исследование стойкости лекарственных препаратов с производственной точки зрения охватывает две проблемы:
- контроль стойкости препаратов, находящихся в аптечной сети;
- контроль стойкости лекарственных средств, технология производства которых находится в стадии разработки.
Архивные образцы должны храниться в заводской упаковке для выяснения влияния атмосферных факторов на препарат, а также проникновения микроорганизмов и потери летучих компонентов препарата. По истечении 2-3 месяцев хранения необходимо провести визуальное наблюдение за внешним видом, беря по одной упаковке препарата (целостность упаковки, внешний вид препарата - цвет, консистенция, образование осадка), а также проверить вес упаковки. Если не наблюдается внешних изменений, то необходимо провести определение активных веществ. Для этого достаточны методы, указанные в стандартах. Если визуально заметны изменения, то необходимо провести исследования при помощи аналитического метода, соответственно подобранного для данного препарата. В жидких лекарственных формах, если выпал осадок, рекомендуется, кроме определения содержания биологически активных веществ в жидкости, полученной после фильтрования препарата, щэоизвести идентификацию образовавшегося осадка. Это возможно только в том случае, если имеется достаточное количество препарата. Осадок, отделенный центрифугированием, следует подвергнуть микроскопическому исследованию (характер осадка), проверить его растворимость, температуру плавления и провести хроматографическое разделение для определения вго однородности.
В случае обнаружения визуальных изменений в одной упаковке, необходимо проверить все остальные упаковки данной серии архива ных образцов для выяснения того, имеем ли мы дело с единичным случаем или с изменениями, наступившими во всех упаковках. Если изменения наблюдаются только спорадически, следует обратить особое внимание на состояние упаковки, ее герметичность и степень наполнения. Если упаковка представляет собой металлическую банку или какой-либо непрозрачный герметический сосуд, без разрушения которого невозможно ознакомиться с состоянием его содержимого, то следует из всей хранимой партии выбрать методом случайного отбора 5-6 упаковок и проанализировать их.
Выводы относительно стойкости препарата зависят от того, до какой степени обнаруженные изменения коснулись его потребительной ценности. Что касается сухих экстрактов, то повышенная влажность их и комковатость делают их непригодными к употреблению. Однако в случае настоек и жидких экстрактов даже сильное изменение их цвета не может служить основанием для их браковки, если этому не сопутствует разложение биологически активных веществ.
Определение стойкости лекарства, технология производства которого еще разрабатывается, является более сложной задачей. Необходимо, с одной стороны, быстро учесть, какое направление могут принять изменения препарата и какие факторы могут их обуславливается, с другой стороны, как можно скорее разработать технологические приемы, обеспечивающие максимальную стойкость препарата.
На крупных современных предприятиях, производящих фармацевтические средства, организация аналитических лабораторий обеспечивает возможность всестсронних исследований по определению стойкости лекарственных препаратов (см. приведенную ниже схему).
Схема организации аналитических лабораторий на современных фармацевтических предприятиях.
Отдельные аналитические лаборатории имеют строго определенные задания. В первой лаборатории разрабатываются новые методы анализа. Здесь технолог получает ответ на вопрос, может ли быть сделан анализ нового препарата (методы, приборы, трудоемкость). В рамках своих основных исследований эта лаборатория занимается изучением процессов разложения препаратов и отождествлением продуктов разложения. Как известно, многие фирмы весьма заинтересованы в возможности проведения сравнительного анализа конкурирующих препаратов. Для этого отведено место в программе работ лaбоpaтopии.
Механические методы искусственного старения (ускоренного) без учета их влияния на специфические свойства препарата неприменимы. Результаты можно считать абсолютно достоверными, если во время испытания не изменяются состав препарата, рН раствора, если речь идет об однородном препарате и если мы уверены, что. процесс разложения при повышенной температуре происходит таким же образом, как и при комнатной. Учитывая тот факт, что зачастую мы имеем дело о двумя и более одновременными процессами разложения, рекомендуется применять умеренные температуры 20°, 35° и 50°С. Это способствует получению более объективных результатов.
При испытании препаратов сложного состава необходимо проверить, не влияют ли на процесс разложения какие-либо иные, параллельно развивающиеся химические или физические процессы. Такие факторы, как изменение рН раствора, выделение некоторых компонентов смеси в виде осадка и др., могут сильно отразиться на ускорении или, наоборот, на замедлении процесса разложения. Тем не менее можно отметить, что при наблюдении происходящих изменений результаты, получаемые при применении метода искусственного старения, можно принять в качестве солидного основания прогнозирования устойчивости препарата. Рекомендуется применять систему продолжительных наблюдений процесса разложения при комнатной температуре как критерия проверки результатов метода ускоренного старения. Вместе с тем, учитывая, что входящие в состав лекарственной формы вспомогательные вещества могут оказывать собственное влияние на процесс разложения, необходимо изучать возможность применения своеобразного метода испытания для каждой лекарственной формы препарата.
УДК 615.45
ИССЛЕДОВАНИЕ СТОЙКОСТИ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ
Проф., д-р Е.ЛЮТОМСКИЙ
Институт растительно-лекарственной промышленности, г. Познань.Проф., д-р Е.ЛЮТОМСКИЙ
Жидкие лекарства. Жидкие лекарства, представляя собой растворы индивидуальных и очищенных биологически активных- веществ, могут исследоваться аналогично препаратам синтетического происхождения, главным образом на основе кинетического метода.
Впрыскиватели. Инъекционные растворы. В виде инъекционных растворов в основном применяются препараты растительного происхождения, максимально очищенные. Однако и они содержат значительное количество балластных веществен на практике надо всегда считаться с их влиянием на процесс разложения. При исследовании стойкости инъекционных растворов следует провести предварительно проверку относительно того, не вызывает ли термическая стерилизация разложения действующего вещества и как происходит разложение вещества в стерильном и нестерильном растворе. Не следует проводить испытания ускоренного старения ампул, которые после стерилизации хранились неопределенное время при комнатной температуре. Кинетические вычисления в таких условиях теряют всякий смысл.
При исследовании инъекционных растворов следует поступать следующим образом:
- Стерилизовать (120° - 20 мин., 110° - 30 мин. либо 100° - 60 мин.) и проверять, разлагается ли активное вещество и в какой степени.
- Провести в условиях ускоренного старения исследования стерильных и нестерильных pacтвоpoв. После получения постоянных результатов можно определить стойкость растворов и сделать выводы о том, стойкий ли исследуемый раствор после термической стерилизации и не следует ли применить другой вид стерилизации, например путем фильтрации.
- Проверить влияние величины рН на ход реакции разложения:
а) проверить, изменяется ли рН во время хранения при комнатной и повышенной температуре;
б) провести исследования в ускоренных условиях с буферными растворами с различными рН (например, от 3 до 9). - Составить график зависимости постоянной К от рН раствора.
- При практических решениях вопроса необходимо иметь в виду то, что составными ингредиентами инъекционных растворов не могут быть буферные растворы, имеющие в своем составе вредные для здоровья вещества, например борная кислота (токсичная), лимонная кислота (уменьшает свертываемость крови за счет выпадения в осадок ионов кальция).
- На ход этой реакции может оказывать влияние и сам буферный раствор. Могут наступать так называемые солевые эффекты действия от так называемой ионной силы. Pacтворы препаратов растительного происхождения часто имеют свой определенный рН,и для получения нужного рН pacтвоpa необходимо провести исследования с целью определения качества и количества буферного вещества необходимо тщательно контролировать рН раствора сразу после его приготовления, а также во время эксперимента. Добавление буферующих веществ не всегда гарантирует поддерживание предусмотренного рН раствора.
- Если раствор в ампуле окисляется, следует провести исследования его стойкости также вне кислородной среды, т.е. исследовать стойкость раствора в ампулах, наполненных нейтральным газом. Кроме того, следует учитывать, что, наряду с разложением веществ под воздействием кислорода, может параллельно проходить другая реакция без участия кислорода. Такой процесс в бескислородных условиях будет проходить беспрепятственно. Обе эти реакции проходят с различной скоростью и имеют свои оптимальные скорости при различных значениях рН. Если разрушение вещества происходит вследствие реакции типа red-ох, следует провести испытания стойкости раствора с добавлением противоокислителей.
Трудности в оценке стойкости растительных экстрактов усугубляются еще и тем, что отсутствуют однородные методы анализа основных веществ. Жидкие лекарственные формы, состав которых может представлять хорошую питательную среду для микрорганизмов (сок, сироп и т.д.), должны подвергаться микробиологической оценке с целью проверки консервирующего действия.
Исследование устойчивости лекарственных форм препаратов растительного происхождения является обычно более трудным, чем исследование инъекционных растворов. Это связано с тем, что в данном случае мы имеем дело с растворами менее очищенными, имеющими в своем составе сопутствующие вещества. Это преимущественно более неустойчивые физико-химические системы. Следует также обратить внимание на изменение окраски осадка. Изменение рН во время проведения исследования ускоренного старения может быть большее,- чем в инъекционных растворах. Из этого следует, что необходимо больше уделять внимания наблюдению за этим параметром. Эти процессы не влияют на точность измерений кинетики процесса разложения активных веществ. Обозначение устойчивости препарата путем экстраполяции имеет в данном случае сугубо ориентировочное значение. Главным моментом являются результаты сравнения исследований устойчивости препаратов в складских условиях и при температуре 35° С.
Спиртовые растворы ясследуются таким же образом. В присутствии спирта электродитъг, и прежде всего кислоты, диссоцируют слабее, что в общем увеличивает их устойчивость.
Твердые лекарственные формы. Порошки принадлежат к самым устойчивым лекарственным формам. Только особо чувствительные вещества могут окисляться кислородом воздуха.
Таблетки и пилюли менее устойчивы. Эти лекарственные фермы всегда содержат какое-то колкчество воды, что не исключает возможности гидролиза. В их состав могут входить стимулнрующие и ускоряющие процесс окисления вещества, как например, перекиси в жирах, жирных кислотах (стеарин) и их солях, ферменты, оксидация (в аравийской камеди).
Проведя испытания устойчивости, следует обратить внимание на то, что процесс влажной грануляции является идеальным условием для разложения и что эта реакция проходит быстрыми темпами. В связи с этим для испытаний следует брать свежеприготовленные таблетки.
Подвергая испытаниям ускоренного старения таблетки спустя несколько месяцев после их изготовления, можно прийти к ошибочным выводам.
Таблетки, драже и пилюди, подвергаемые ускоренному старению при повышенной температуре, надо поместить в камерах с относительной влажностью до 80% с целью определения чувствительности к действию температуры (подбор упаковочного материала). После проведения испытаний по ускоренному старению эту группу препаратов необходимо исследовать на распад и быстроту растворения активных веществ. Кроме этого, следует провести проверку на прочность таблеток и герметичность оболочки таблеток-драже.
Следует отметить, что оболочка защищает таблетки только в некоторой степени от влияния кислорода и влаги воздуха. Однако эта защита не всегда бывает эффективной. Некоторые составные части оболочки мoгут даже ускорять процесс разложения, а сам процесс приготовления таблеток, покрытых оболочкой, может оказывать отрицательное влияние.
Постоянные реакции в случае таблеток и драже имеют большие отклонения, чем в растворах, где химические реакции проходят более равномерно. Поэтому устойчивость таблеток и драже следует определять на основе результатов анализов (в промежутках через 3, 6 и 12 месяцев) в условиях складской температуры, т.е. при 20°С. За исключением таблеток с углекислым натрием, которые целесообразно проверять при температуре 35° С.
Гораздо большие трудности доставляют растительные сборы и гранулированные формы, устойчивость которых зависит не только от условий хранения, но прежде всего от упаковочного материала.
Мягкие лекарственные формы. Мази и суппозитории, имеющие мягкую консистенцию, подвергаются изменениям во время хранения при повышенных температурах, вследствие чего вюжет возникать седнментаджаг взвешенных частиц. При этом контрольный анализ, если не приведет к первоначальной гомогенности препарата, может показать не истинный результат.
Нельзя применять повышенные температуры для ускоренного старения мазей и суппозиториев. Контроль стойкости рекомендуется проводить при температурах 20° и 30° С без кинетических исследований. Мази, не содержащие в своем составе воду, довольно надежно сохраняют действуюпще вещества от внешнего воздействия. Вредное влияние на устойчивость лекарственных форм может оказывать разложение самой основы - прежде всего перекиси. Эти вещества могут быть найдены во всех основах мазей, особенно в ланолине и жире. Основы необходимо тщательно исследовать на присутствие перекисей. Мази, содержащие воду, менее стойкие. Линименты типа о/W имеют такую же устойчивость приблизительно, как водяные растворы, и разрушение их можно предвидеть из достоянной К, полученной путем ускоренного старения водных растворов.
Исследование устойчивости линиментов в условиях усксренного старения затруднительно, хак как подогретый линимент расслаивается. Исследовать устойчивость линиментов можно только при комнатной температуре и подкреплять эти исследования ориентировочными данными ускоренного старения действующего вещества в основах. Кроме того, следует проверить, не ухудшается ли структура линимента, проверяя ее реологические свойства.
Суппозитории. Исследование устойчивости суппозиториев производится аналогично исследованиям мази. Вредным является также содержание перекисей в основах. Однако основания суппозиториев, как правило, более стойки, чем основы мазей.
Расходы, связанные с исследованиями устойчивости лекарственных веществ, довольно высоки, и поэтому результат экономического анализа должен всякий раз определять целееоофазность проведения подобных исследований в производственных условиях. В связи с большой физиологической активностью расширенные исследования должны проводиться в первую очередь с препаратами, в состав которых входят гликозиды и алкалоиды.
Проблема устойчивости лекарственных средств охватывает очень широкий круг вопросов, начиная с аналитической химии, технологии лекарственных форм, физической химии; кончая их хранением и юридическими вопросами.
Возможны расхождения технических характеристик товара при продаже в
зависимости от модификации и страны происхождения. Возможны текстовые
опечатки.
Мы не несем никакой ответственности за результаты применения
приведенных рецептов или методов лечения. Есть противопоказания. Не
занимайтесь самолечением. Посоветуйтесь с врачом.
Наш сайт не является публичной офертой, определяемой положениями Статьи 437 (2) ГК РФ., а носит исключительно информационный характер. Для получения точной информации о наличии и стоимости товара, пожалуйста, обращайтесь по нашим телефонам. В случае копирования, использования любого материала находящегося на сайте www.lekarstvennye-rasteniya.ru, активная ссылка обязательна, в случае печати – печатная ссылка. Копирование структуры сайта, идей или элементов дизайна сайта строго запрещено. Права на все торговые марки, изображения и материалы, представленные на сайте, принадлежат их владельцам.
